
Das Joubert-Syndrom ist eine Störung genetischen Ursprungs, die durch eine Abnahme des Muskeltonus, Koordinationsprobleme, abnormale Augenbewegungen, veränderte Atmungsmuster und geistige Behinderung gekennzeichnet ist (Joubert Syndrome Foundation, 2016)..
Alle diese Veränderungen sind auf eine autosomale genetische Übertragung zurückzuführen, die wichtige Gehirnanomalien, eine Verringerung des Kleinhirnwurms sowie Anomalien in der Struktur des Hirnstamms verursacht (Nationales Institut für neurologische Störungen und Schlaganfall, 2016)..
Darüber hinaus gehört das Joubert-Syndrom zu einer Gruppe von Erkrankungen, die als Ciliopathien bezeichnet werden und eine Funktionsstörung eines Teils der Zellen betreffen, die als Zilien bezeichnet werden. Joubert-Syndrom-Stiftung, 2016).
Die Erstbeschreibung dieser Pathologie erfolgte 1968 durch Marie Joubert et al., In der vier Fälle beschrieben wurden. Bei den Patienten gab es eine teilweise oder vollständige Abwesenheit des Kleinhirnwurms, des episodischen Ampnoe-Hypernea-Syndroms bei Neugeborenen, abnormaler Augenbewegungen, Ataxie und geistiger Behinderung (Angemi und Zucotti, 2012)..
Darüber hinaus war dieses Syndrom auch mit verschiedenen Multiorganveränderungen verbunden, wie Leberfibrose, Polydaktylie, Nephronoptyse oder Netzhautdystrophie (Angemi und Zucotti, 2012)..
In Bezug auf die Behandlung gibt es derzeit keine Heilung für das Joubert-Syndrom. Therapeutische Interventionen zielen auf symptomatische Kontrolle und Unterstützung, körperliche und geistige Stimulation von Kindern und Ergotherapie ab (Nationales Institut für neurologische Störungen und Schlaganfall, 2016)..
Artikelverzeichnis
Das Joubert-Syndrom (JS) ist eine Art von Pathologie genetischen Ursprungs, die durch eine angeborene Fehlbildung in den Bereichen des Hirnstamms und der Genese (teilweise oder vollständige Abwesenheit) oder Hypoplasie (unvollständige Entwicklung) des Kleinhirnwurms gekennzeichnet ist, die (Ophatnet) verursachen kann , 2009).
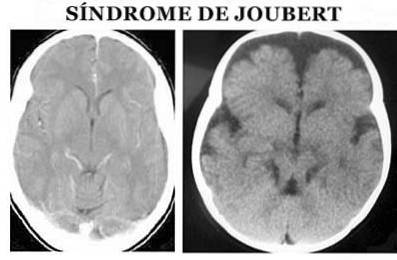
Insbesondere auf anatomischer Ebene ist es durch das sogenannte molare Zeichen des Mittelhirns gekennzeichnet: Agenese oder Hypoplasie des Kleinhirnwurms, Verengung der oberen Kleinhirnstiele mit Verdickung, Dehnung und fehlender Diskussion und tiefe Fossa interpeduncularis (Angemi und Zuccoti, 2012).
Es handelt sich um eine Störung, die viele Bereiche und Organe des Körpers betreffen kann, sodass die Anzeichen und Symptome bei den Betroffenen erheblich variieren (US National Library of Medicine, 2011)..
Die meisten Betroffenen leiden unter einem geschwächten Muskeltonus (Hypotonie) und motorischen Koordinationsschwierigkeiten (Ataxie). Weitere charakteristische Merkmale sind: Episoden veränderter Atmung, Nystagmus (unwillkürliche und arrhythmische Bewegung der Augen), verzögerte motorische Entwicklung und variable intellektuelle Schwierigkeiten (US National Library of Medicine, 2011)..
Die Prävalenz des Joubert-Syndroms wurde auf ungefähr 1 / 80.000 bis 1 / 100.000.000 Lebendgeburten geschätzt. Weltweit wurden mehr als 200 klinische Fälle registriert (Angemi und Zuccoti, 2012).
Viele Fachleute halten diese Zahlen für unterschätzt, da das Joubert-Syndrom eine Vielzahl von Erkrankungen aufweist und weitgehend unterdiagnostiziert ist (US National Library of Medicine, 2011)..
Viele der klinischen Symptome des Joubert-Syndroms sind im Kindesalter mehr als offensichtlich. Viele betroffene Kinder haben erhebliche motorische Verzögerungen (National Organization for Rare Disease, 2011)..
Die häufigsten Merkmale des klinischen Verlaufs sind: mangelnde Muskelkontrolle (Ataxie), veränderte Atmungsmuster (Hyperkapnie), Schlafapnoe, abnormale Augenbewegungen (Nystagmus) und niedriger Muskeltonus (National Organization for Rare Disease, 2011)..
Andererseits umfassen einige der Veränderungen, die mit dem Joubert-Syndrom verbunden sein können, unter anderem: veränderte Entwicklung der Netzhaut, Abnormalitäten in der Iris, Strabismus, Nieren- und / oder Leberveränderungen, Vorsprung der Membranen, die das Gehirn bedecken ( Nationale Organisation für seltene Krankheiten, 2011).
Alle von diesem Syndrom abgeleiteten Veränderungen umfassen verschiedene Bereiche: neurologische, okulare, renale und muskuloskelettale Veränderungen (Bracanti et al., 2010).
Die charakteristischsten neurologischen Veränderungen des Joubert-Syndroms sind Bracanti et al., 2010): Hypotonie, Ataxie, allgemeine Entwicklungsverzögerung, intellektuelle Veränderungen, Veränderung der Atmungsmuster und abnorme Augenbewegungen.
Auf körperlicher Ebene ist die Netzhaut eines der vom Joubert-Syndrom betroffenen Organe. Veränderungen in diesem Organ treten in Form einer Netzhautdystrophie aufgrund einer fortschreitenden Degeneration der für den Fotoempfang verantwortlichen Zellen auf.
Klinisch können Augenveränderungen von angeborener Netzhautblindheit bis hin zu fortschreitender Netzhautdegeneration reichen.
Andererseits ist es auch möglich, das Vorhandensein des Koloboms zu beobachten. Diese Augenveränderung ist ein angeborener Defekt, der die Augeniris betrifft und als Loch oder Schlitz erscheint.
Nierenerkrankungen betreffen mehr als 25% der vom Joubert-Syndrom Betroffenen.
In vielen Fällen können Nierenanomalien mehrere Jahre lang asymptomatisch bleiben oder sich mit unspezifischen Anzeichen manifestieren, bis sie sich als akutes oder chronisches Nierenversagen darstellen..
Nach den ersten Beschreibungen dieser Pathologie ist Polydaktialie (eine genetische Störung, die die Anzahl der Finger oder Zehen erhöht) ein häufiger klinischer Befund..
Darüber hinaus ist es auch üblich, orofaziale oder strukturelle Anomalien auf der Ebene der Wirbelsäule zu beobachten.
Experimentelle Studien haben das Joubert-Syndrom als autosomal-rezessive Störung eingestuft (National Organization for Rare Disease, 2011)..
Eine autosomal rezessive genetische Störung bedeutet, dass zwei Kopien eines abnormalen Gens vorhanden sein müssen, damit das Merkmal oder die Krankheit vorliegt (National Institutes of Health, 2014)..
Daher tritt eine rezessive genetische Veränderung auf, wenn eine Person von jedem Elternteil dasselbe abnormale Gen für dasselbe Merkmal erbt. Wenn eine Person nur eine Kopie des mit der Krankheit verbundenen Gens erhält, ist sie Trägerin, zeigt jedoch keine Symptome (National Organization for Rare Disease, 2011)..
Darüber hinaus wurden mindestens zehn Gene als eine der möglichen Ursachen des Joubert-Syndroms identifiziert (National Organization for Rare Disease, 2011)..
Eine Mutation im AHI1-Gen ist in etwa 11% der betroffenen Familien für diesen pathologischen Zustand verantwortlich. Bei Menschen mit dieser genetischen Veränderung sind Sehveränderungen aufgrund der Entwicklung einer Netzhautdystrophie häufig (Nationale Organisation für seltene Krankheiten, 2011)..
Die Mutation des nphp1-Gens ist die Ursache für ungefähr 1-2% der Fälle des Joubert-Syndroms. Bei Personen mit dieser genetischen Veränderung sind Nierenveränderungen häufig (National Organization for Rare Disease, 2011)..
Andererseits ist eine CEP290-Genmutation die Ursache für 4-10% der Fälle des Joubert-Syndroms (National Organization for Rare Disease, 2011)..
Darüber hinaus hängen Mutationen in den Genen TME67, JBTS1, JBTS2, JBTS7, JBTS8 und JBTS9 auch mit der Entwicklung des Joubert-Syndroms zusammen (National Organization for Rare Disease, 2011)..
Die Diagnose des Joubert-Syndroms wird anhand der körperlichen Symptome gestellt. Es ist notwendig, sowohl eine detaillierte körperliche Untersuchung als auch die Verwendung verschiedener diagnostischer Tests, insbesondere Magnetresonanzbilder, durchzuführen (Ophatnet, 2009)..
Darüber hinaus werden molekulargenetische Tests häufig verwendet, um genetische Veränderungen zu identifizieren, die in 40% der Fälle des Joubert-Syndroms nachgewiesen wurden (National Organization for Rare Disease, 2011)..
Andererseits ist es auch möglich, eine pränatale Diagnose dieser Pathologie durch fetalen Ultraschall und molekulare Analyse zu stellen, insbesondere in Familien mit einer genetischen Vorgeschichte des Joubert-Syndroms (Ophatnet, 2009)..
Wenn die charakteristischsten Merkmale des Joubert-Syndroms in Kombination mit einer oder mehreren zusätzlichen physischen Pathologien auftreten, kann eine Diagnose des Joubert-Syndroms und verwandter Störungen (JSRD) gestellt werden (US National Library of Medicine, 2011)..
Abhängig von der Art der verwandten Pathologie, die mit dem Vorhandensein des Joubert-Syndroms verbunden ist, können wir daher Subtypen davon finden. Das Klassifizierungssystem für das Joubert-Syndrom befindet sich jedoch aufgrund der Entdeckung genetischer Beiträge und der besseren Kenntnis der phänotypischen Korrelationen noch in einer Evolutionsphase..
Wir können daher finden (Bracanti et al., 2010):
Die Behandlung des Joubert-Syndroms ist symptomatisch und unterstützt die zugrunde liegenden Pathologien. Zusätzlich zu pharmakologischen Interventionen ist es üblich, eine frühe körperliche und kognitive Stimulation anzuwenden (Nationales Institut für neurologische Störungen und Stoke, 2016)..
Wenn Atemwegsveränderungen, insbesondere in den Anfangsphasen des Lebens, signifikant sind, muss die Atemfunktion überwacht werden (National Institute of Neurological Disorders and Stoke, 2016)..
Andererseits sollte die Identifizierung und Kontrolle von Augendegeneration, Nierenkomplikationen und den restlichen Komplikationen im Zusammenhang mit dem Joubert-Syndrom so früh wie möglich durchgeführt werden, um die therapeutischen Maßnahmen anzupassen (Nationales Institut für neurologische Störungen und Stoke, 2016) ).



Bisher hat noch niemand einen Kommentar zu diesem Artikel abgegeben.