Das Phakomatose ist eine Gruppe von neurokutanen Störungen genetischen Ursprungs, die in der Allgemeinbevölkerung selten sind. Auf klinischer Ebene sind sie durch die Entwicklung einer multisystemischen organischen Beteiligung an Haut- oder Tumorläsionen in verschiedenen Bereichen der Haut, der Organe oder des Nervensystems gekennzeichnet..
Darüber hinaus erschwert der unspezifische klinische Verlauf die frühzeitige Diagnose, sodass die medizinischen und psychologischen Folgen die Lebensqualität der betroffenen Person und ihrer Angehörigen erheblich beeinträchtigen..
Obwohl es eine große Anzahl von neurokutanen Erkrankungen gibt, gehören zu den häufigsten Fibromatosen vom Typ I und Typ II, die Bourneville-Krankheit, das Sturge-Weber-Syndrom und die Von-Hippel-Lindau-Krankheit..
Andererseits wurden trotz der Tatsache, dass dies alles angeborene Pathologien sind, mehrere therapeutische Ansätze dermatologischer Natur entwickelt, die versuchen, die für diese Störungen charakteristischen Anzeichen und Symptome und damit die medizinische Prognose der Betroffenen zu verbessern.
Artikelverzeichnis
Der Begriff Phakomatose stammt aus dem Ausdruck griechischen Ursprungs Phakos deren Bedeutung bezieht sich auf <
Neurokutane Pathologien sind grundsätzlich durch das Bestehen eines signifikanten Zusammenhangs zwischen einer neurologischen Beeinträchtigung oder Störung und den dermatologischen Manifestationen gekennzeichnet.
Daher wird der Begriff neurokutane Pathologie allgemein verwendet, um verschiedene Krankheiten zu erfassen, die bei der angeborenen betroffenen Person vorhanden sind und die zusätzlich während des gesamten Lebens mit der Entwicklung von Hautläsionen und Tumoren in verschiedenen Bereichen, dem Nervensystem, auftreten können. Herz-Kreislauf-System, Nierensystem, Hautsystem, Augensystem usw..
Auf diese Weise wurde der Begriff Phakomatose 1917 von Brouwer und später von van der Hoeve 1923 eingeführt. In den ersten Beschreibungen wurde jedoch nur auf einige in dieser Gruppe enthaltene Pathologien Bezug genommen. Derzeit mehr als 40.
Auf klinischer Ebene wird Phakomatose als eine Krankheit beschrieben, die Hautveränderungen und gutartige / bösartige Fehlbildungen in verschiedenen Systemen aufweist: neurologisch, okular, kutan und viszeral..
In Bezug auf die betroffenen Bereiche weisen verschiedene Autoren darauf hin, dass diejenigen ektodermalen Ursprungs am stärksten betroffen sind, dh die Haut und das Nervensystem, obwohl sie auch andere Systeme oder Geräte wie das Auge betreffen können.
Syndrome und Pathologien neurokutanen Ursprungs sind seltene Krankheiten in der Allgemeinbevölkerung, obwohl es zu all diesen keine spezifischen Daten auf allgemeiner Ebene gibt.
Daher variiert die Epidemiologie dieser Störungen in Abhängigkeit von der Art der Krankheit, insbesondere ist die Neurofibromatose eine der häufigsten mit einer relativen Prävalenz von einem Fall pro 300.000 Geburten.
Neurokutane Erkrankungen sind durch die Entwicklung von Hautläsionen gekennzeichnet. Insbesondere unterscheidet sich die Phakomatose von vielen anderen durch das Vorhandensein von Hamartomen..
Hamartome sind eine Art von gutartigen Missbildungen oder Tumoren, die in verschiedenen Organen wie Gehirn, Herz, Augen, Haut oder Lunge wachsen können..
Phakomatose kann jedoch mit einer Vielzahl von Erkrankungen verbunden sein, die je nach der spezifischen Krankheit oder Pathologie der betroffenen Person grundlegend variieren..
Gegenwärtig wurde eine große Anzahl von neurokutanen Störungen auf klinischer und genetischer Ebene identifiziert, es gibt jedoch einige mit einer höheren Prävalenz in der Allgemeinbevölkerung: Neurofibromatose Typ I und Typ II, Bourneville-Krankheit, Von-Krankheit Hippel-Lindau und Sturge-Weber Syndrom.
Es gibt verschiedene klinische Formen der Neurofibromatose. Gegenwärtig sind jedoch die Typ-I-Neurofibromatose, auch Von Reclinghausen-Krankheit genannt, und die Typ-II-Neurofibromatose, gefolgt von einer spinalen Shwannomatose, am häufigsten.
Auf ätiologischer Ebene haben alle diese medizinischen Manifestationen der Neurofibromatose einen genetischen Ursprung und treten bei der Bildung von Tumoren in Nervenbereichen auf, insbesondere im zentralen und peripheren Nervensystem..
Tumorformationen, normalerweise nicht krebsartig oder gutartig, neigen dazu, fast überall im Nervensystem zu wachsen und sich zu entwickeln, wie z. B. im Gehirn, im Rückenmark oder in peripheren Nerven.
Zu den Algen sekundärer medizinischer Komplikationen bei der Neurofibromatose gehören unter anderem Wachstumsstörungen, die Entwicklung von Anfällen, das Auftreten von Hirntumoren, Knochenerkrankungen, Taubheit und / oder Blindheit oder die Entwicklung signifikanter Lernprobleme.
Darüber hinaus ist diese Pathologie vom Moment der Geburt an vorhanden. Die signifikante Manifestation seines Krankheitsbildes kann sich jedoch bis zum späten Säuglingsalter, frühen Jugendalter oder Erwachsenenalter verzögern..
Andererseits umfasst die Diagnose dieser Art von Pathologie neben der physischen und neurologischen Untersuchung üblicherweise verschiedene Neuroimaging-Tests und genetische Analysen..
Darüber hinaus gibt es derzeit keine Heilung für Neurofibromatose. Es gibt jedoch spezielle therapeutische Ansätze zur Kontrolle der dermatologischen Beeinträchtigung. Sie können sowohl pharmakologische als auch chirurgische Behandlungen umfassen, um Tumorbildungen zu stoppen oder zu beseitigen.
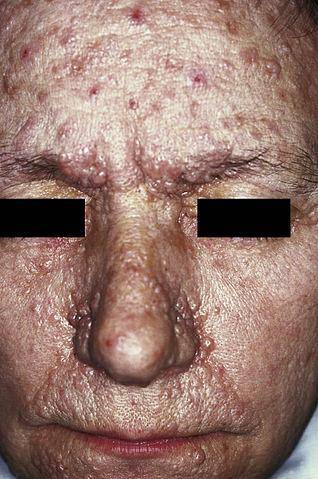
Die Neurofibromatose Typ I (NF1), auch bekannt als von Recklinghausen-Krankheit, äußert sich hauptsächlich in hellbraunen Flecken, die üblicherweise als „Café au lait“, Epheliden (Sommersprossen) und Neurofibrome (Nervenschäden in Schwannschen Zellen und Neuriten) bezeichnet werden..
Es hat einen autosomal dominanten genetischen Ursprung, insbesondere aufgrund einer Mutation auf Chromosom 17 an Position 17q11.2. Somit ist das Gen an
Die Entwicklung der Typ I-Neurofibromatose spielt eine herausragende Rolle bei der Modulation des Zellwachstums und der Zelldifferenzierung und kann darüber hinaus als Tumorsuppressor fungieren.
In Bezug auf die Epidemiologie dieser Pathologie wird eine ungefähre Prävalenz von einem Fall pro 2.500.3000 Geburten angegeben.
Die Diagnose einer Typ-I-Neurofibromatose wird normalerweise auf der Grundlage der konsensklinischen Kriterien des National Institute of Health (1987) gestellt. Sie erfordert jedoch eine kontinuierliche Nachverfolgung, um sekundäre medizinische Komplikationen zu vermeiden..
Normalerweise werden Tumorwachstum mit Medikamenten behandelt, um ihre exponentielle Entwicklung zu verhindern, oder durch chirurgische Entfernung.
Neurofibromatose Typ II (NF2) manifestiert sich hauptsächlich durch die Entwicklung von Schwannomen, dh Tumorbildungen, die von Shcwaan-Zellen stammen und für die Abdeckung der Nervenverlängerungen verantwortlich sind.
Schwannome oder Neuriome betreffen in der Regel insbesondere den Hör- und Sehnerv und in geringerem Maße die Hautpartien.
Typ-II-Neurofibromatose hat einen autosomal dominanten genetischen Ursprung, insbesondere aufgrund des Vorhandenseins einer Mutation auf Chromosom 22 an Position 22q11.22.
Das Gen, das an der Entwicklung dieser Pathologie beteiligt ist, ist für die Kodierung einer Proteinkomponente verantwortlich, die eine herausragende Rolle bei der Tumorsuppression spielt, so dass seine mangelnde Aktivität zu einer abnormalen Zunahme der Zellproliferation führt.
In Bezug auf die Epidemiologie dieser Pathologie ist sie weniger häufig als Typ 1 und weist eine ungefähre Prävalenz von einem Fall pro 50.000 Geburten auf.
Die Diagnose einer Typ-II-Neurofibromatose ähnelt der des vorherigen Typs und wird normalerweise auf der Grundlage der konsensklinischen Kriterien des National Institute of Health gestellt. In der Regel umfasst es jedoch ergänzende Labortests, z. B. Neuroimaging..
Normalerweise werden Tumorwachstum mit Medikamenten behandelt, in Fällen, in denen dies möglich ist, wird jedoch eine chirurgische Entfernung angewendet.
Die Bourneville-Krankheit ist einer der Begriffe, die für Tuberkulose verwendet werden, eine genetische Störung, die durch das Vorhandensein von Hamartomen gekennzeichnet ist.
Auf klinischer Ebene kann es zu einer multisystemischen Erkrankung kommen, die durch Hautstörungen (Gesichtsangiome, Nagelfibrome, fibröse Plaques, hypochrome Flecken usw.), Nierenerkrankungen (Nierenangiomyolipome oder Nierenzysten), Herzerkrankungen (Herzrhabdomyome) gekennzeichnet ist. , neurologische Beeinträchtigung (kortikale Knollen, subependymale Gliazellenknoten, Atrozytome, Anfälle, geistige Behinderung, Verhaltens- und motorische Anomalien), unter anderem.
Wie bei den oben beschriebenen Krankheiten ist der Ursprung der Tuberkulose genetisch bedingt. Insbesondere ist es auf das Vorhandensein von Mutationen in den TSC1- und TSC2-Genen zurückzuführen..
Andererseits wird die Diagnose der Tuberkulose auf der Grundlage der klinischen Kriterien gestellt, die 1998 auf einer medizinischen Konferenz vorgeschlagen wurden. Die genetische Studie wird jedoch auch als relevant für ihre Bestätigung angesehen.
In Bezug auf die Behandlung von Tuberkulose, obwohl es keine Heilung gibt, werden normalerweise verschiedene pharmakologische und chirurgische Ansätze verwendet, hauptsächlich zur Kontrolle des Tumorwachstums und sekundärer medizinischer Komplikationen wie neurologischer Manifestationen.
Die Von-Hippel-Lindau-Krankheit, auch als Retino-Kleinhirn-Angiomatose bekannt, manifestiert sich hauptsächlich durch das Vorhandensein und die Entwicklung von Gefäßfehlbildungen, Zysten und / oder Tumoren, die im Allgemeinen gutartig sind..
Es hat einen autosomal dominanten genetischen Ursprung, insbesondere aufgrund einer Mutation auf Chromosom 3 an Position 3p-25-26. Darüber hinaus wird eine geschätzte Inzidenz von einem Fall pro 40.000 Geburten angegeben..
Insbesondere betrifft die Von-Hippel-Lindau-Krankheit hauptsächlich das Zentralnervensystem (ZNS) und die Netzhaut durch die Bildung von Hämangiomen.
Hämangiome sind Gefäßfehlbildungen, die durch das Vorhandensein von Clustern erweiterter Blutkapillaren gekennzeichnet sind. Sie treten normalerweise im Gehirn- und Wirbelsäulenbereich auf, obwohl sie auch häufig in der Netzhaut oder auf der Haut auftreten.
Die Diagnose dieser Pathologie erfordert zusätzlich zur physischen und neurologischen Untersuchung eine detaillierte ophthalmologische Untersuchung zusammen mit der Analyse aus verschiedenen Neuroimaging-Tests, um das Vorhandensein von Nervenläsionen zu bestätigen..
In Bezug auf die Behandlung der Von-Hippel-Lindau-Krankheit ist die grundlegende Intervention die Operation zur Beseitigung von Gefäßfehlbildungen. Es erfordert jedoch eine kontinuierliche Überwachung, um sekundäre Komplikationen zu vermeiden..
Darüber hinaus hat es eine verringerte Lebenserwartung im Alter von etwa 50 Jahren, hauptsächlich aufgrund der Entwicklung von Nierenzellkarzinomen (neoplastische Formationen von Krebszellen in den Nierentubuli)..
Das Sturge-Weber-Syndrom, auch als Enzephalo-Trigeminus-Angiomatose bekannt, manifestiert sich hauptsächlich durch das Vorhandensein von Hämangiomen.
Ein Hämangiom ist eine Art von Neoplasma oder Tumorbildung, die durch das Vorhandensein einer ungewöhnlich hohen Anzahl von Blutgefäßen in der Haut oder anderen inneren Organen gekennzeichnet ist..
Insbesondere auf klinischer Ebene ist das Sturge-Weber-Syndrom durch die Entwicklung von Gesichtshämangiomen, intrakraniellen Hämangiomen sowie Chorid-, Bindehaut-, Episzeral- und Glaukomhämangiomen gekennzeichnet..
Es hat einen genetischen Ursprung, insbesondere aufgrund einer Mutation auf Chromosom 9 an Position 9q21 im GNQ-Gen. Diese genetische Komponente spielt eine herausragende Rolle bei der Kontrolle von Wachstumsfaktoren, vasoaktiven Peptiden und Neurotransmittern (Orhphanet, 2014)..
Die Diagnose des Sturge-Weber-Syndroms basiert auf klinischem Verdacht und verschiedenen Labortests wie Computertomographie oder Magnetresonanztomographie..
Andererseits ist die Lasertherapie in Bezug auf die Behandlung in der Lage, das Fortschreiten dieser Pathologie zu verringern und darüber hinaus in vielen Fällen Hämangiome vollständig zu eliminieren..

Bisher hat noch niemand einen Kommentar zu diesem Artikel abgegeben.