
Das Williams-Syndrom Es handelt sich um eine Entwicklungsstörung genetischen Ursprungs, die mit einem charakteristischen Profil körperlicher und kognitiver Beeinträchtigungen verbunden ist. Speziell auf klinischer Ebene ist es durch 4 Kardinalpunkte gekennzeichnet: 1) atypische Gesichtsmerkmale und -merkmale, 2) allgemeine Verzögerung der psychomotorischen Entwicklung und des spezifischen neurokognitiven Profils, 3) kardiovaskuläre Veränderungen und t) Möglichkeit der Entwicklung einer kindlichen Hyperkalzämie.
Trotz der Tatsache, dass das Williams-Syndrom als seltene Krankheit gilt, gibt es weltweit Tausende von Betroffenen. In Bezug auf die Diagnose liefert die klinische Untersuchung normalerweise die notwendigen Ergebnisse für ihre Etablierung. Um jedoch andere Pathologien und falsch positive Ergebnisse auszuschließen, wird eine genetische Studie normalerweise mit verschiedenen Techniken gestartet.
Auf der anderen Seite gibt es weder eine Heilung für das Williams-Syndrom noch ein Standardbehandlungsprotokoll, so dass die meisten therapeutischen Interventionen versuchen werden, medizinische Komplikationen zu regulieren. Darüber hinaus müssen Frühförderprogramme, individuelle Sonderpädagogik und neuropsychologische Stimulation in die Interventionen einbezogen werden..
Artikelverzeichnis
Das Williams-Syndrom ist eine Entwicklungsstörung, die verschiedene Bereiche erheblich betreffen kann.
Im Allgemeinen ist diese Pathologie durch das Vorhandensein atypischer Gesichtsmerkmale oder kardiovaskulärer Veränderungen, mäßiger geistiger Behinderung, Lernprobleme und charakteristischer Persönlichkeitsmerkmale gekennzeichnet..
So wurde der erste Patient mit Williams-Syndrom von Dr. Guido Fanconi in einem klinischen Bericht von 1952 beschrieben. Es war jedoch der Kardiologe Joseph Williams, der diese Pathologie 1961 genau identifizierte und von den Deutschen Beuren beschrieben wurde.
Aus diesem Grund erhält das Williams-Syndrom seinen Namen von beiden Autoren (Williams-Beuren-Syndrom) oder einfach vom ersten.
Trotz der Tatsache, dass bis vor einigen Jahren die Identifizierung der Pathologie anhand der phänotypischen Merkmale durchgeführt wurde, fanden Edward et al. 1993 eine genetische Abnormalität in Chromosom 7q 11.23 als ätiologische Ursache..
Obwohl das Williams-Syndrom mit einer Vielzahl von sekundären medizinischen Komplikationen verbunden ist, weist es keine hohe Sterblichkeitsrate auf. In vielen Fällen können Betroffene ein eigenständiges Funktionsniveau erreichen.
Das Williams-Syndrom wird als seltene oder seltene genetische Störung angesehen.
Die Williams-Syndrom-Vereinigung hat unter anderem geschätzt, dass das Williams-Syndrom eine Prävalenz von ungefähr 1 Fall pro 10.000 Menschen weltweit hat. Insbesondere wurde festgestellt, dass in den Vereinigten Staaten etwa 20.000 oder 30.000 betroffen sein können.
In Bezug auf die Verteilung der Pathologie nach Geschlecht gibt es keine aktuellen Daten, die auf eine höhere Prävalenz in einer von ihnen hinweisen. Außerdem wurden keine Unterschiede zwischen geografischen Regionen oder ethnischen Gruppen festgestellt.
Andererseits wissen wir auch, dass das Williams-Syndrom eine sporadische Erkrankung ist, obwohl einige Fälle von Familienübertragung beschrieben wurden.
Das Williams-Syndrom hat wie andere Pathologien genetischen Ursprungs einen klinischen Verlauf, der durch eine Beteiligung mehrerer Systeme gekennzeichnet ist.
Viele Autoren, wie González Fernández und Uyaguari Quezada, beschreiben das klinische Spektrum des Williams-Syndroms, das in verschiedene Bereiche unterteilt ist: biomedizinische Merkmale, psychomotorische und kognitive Merkmale, psychologische und Verhaltensmerkmale unter anderem..
Die beim Wiliams-Syndrom vorhandene körperliche Beeinträchtigung ist vielfältig und gehört zu den häufigsten klinischen Befunden, die wir beobachten können:
Eine verzögerte oder verlangsamte Entwicklung kann bereits während der Schwangerschaft festgestellt werden. Kinder, die vom Williams-Syndrom betroffen sind, werden häufig mit geringem Gewicht und geringer Größe geboren. Sobald das Erwachsenenstadium erreicht ist, ist die Gesamthöhe normalerweise niedriger als die der Allgemeinbevölkerung, ungefähr 10-15 cm.
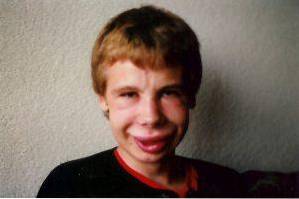
Gesichtsveränderungen sind einer der charakteristischsten klinischen Befunde bei diesem Syndrom. Bei Betroffenen können wir eine deutlich schmale Stirn, ausgeprägte Hautfalten in der Palpebralfissur, Strabismus, Sterniris, kurze und abgeflachte Nase, hervorstehende Wangenknochen und ein kleineres Kinn als gewöhnlich beobachten..
Bei Veränderungen im Zusammenhang mit der Entwicklung von Muskeln und Knochen können unter anderem verminderte Muskeltonus und -stärke, Gelenkschlaffheit, Skoliose und Kontrakturen beobachtet werden. Visuell kann eine Haltung beobachtet werden, die durch herabhängende Schultern und halbgebogene untere Extremitäten gekennzeichnet ist..
Obwohl es normalerweise keine signifikanten Anomalien oder Missbildungen in der Ohrmuschel gibt, entwickelt sich in allen Fällen eine Erhöhung der Hörempfindlichkeit. Betroffene Personen neigen dazu, bestimmte Geräusche als störend oder schmerzhaft wahrzunehmen oder zu erleben.
Die Haut hat normalerweise eine geringe Elastizität, so dass frühe Zeichen der Hautalterung beobachtet werden können. Darüber hinaus können sich Hernien entwickeln, insbesondere im Leisten- und Nabelbereich..
Die verschiedenen Anomalien im Herzen und in den Blutgefäßen stellen die bedeutendste medizinische Komplikation dar, da sie das Überleben der betroffenen Person gefährden können.
Unter den kardiovaskulären Anomalien sind einige der häufigsten die supravalvuläre Aortenstenose, die Lungenaststenose und die Aortenklappenstenose. All diese Veränderungen auf klinischer Ebene können aufgrund der Entwicklung einer arteriellen Hypertonie andere Gefäßgebiete und sogar das Gehirn betreffen.
Anomalien im Zusammenhang mit Nierenfunktion und Blase sind sehr häufig. Darüber hinaus kann auch eine Anreicherung von Kalzium (Nephrokalzinose), Harndrang oder nächtliche Enuresis festgestellt werden.
Auf der kognitiven Ebene bestehen die wichtigsten Merkmale in einer allgemeinen Verzögerung beim Erwerb motorischer Fähigkeiten, einer moderaten intellektuellen Verzögerung und verschiedenen Veränderungen im Zusammenhang mit der visuellen Wahrnehmung.
Es werden verschiedene Veränderungen im Zusammenhang mit Gleichgewichts- und Koordinationsproblemen beschrieben, die hauptsächlich auf das Vorhandensein von Anomalien des Bewegungsapparates zurückzuführen sind und unter anderem zu einer Verzögerung des Gangerwerbs, der endgültigen motorischen Fähigkeiten usw. führen..
Es ist möglich, eine mäßige geistige Behinderung zu finden, der typische IQ der Betroffenen schwankt normalerweise zwischen 60 und 70. In Bezug auf die spezifischen betroffenen Bereiche besteht eine deutliche Asymmetrie: Zusätzlich zur psychomotorischen Koordination, Wahrnehmung und visuellen Integration ist dies normalerweise der Fall deutlich betroffen sein, während Bereiche wie die Sprache in der Regel weiter entwickelt sind.
In den ersten Phasen kommt es normalerweise zu einer Verzögerung beim Erwerb von Sprachkenntnissen, die sich jedoch in der Regel um 3-4 Jahre erholt. Kinder mit Williams-Syndrom haben in der Regel eine gute Ausdruckskommunikation, können kontextualisiertes Vokabular, korrekte Grammatik, Augenkontakt, Gesichtsausdrücke usw. verwenden..
Einer der wichtigsten Befunde beim Williams-Syndrom ist das außergewöhnliche Sozialverhalten der Betroffenen. Obwohl in einigen Fällen Angstkrisen oder übermäßige Sorgen auftreten können, sind sie sehr einfühlsam und sensibel.
Die jüngsten Untersuchungen haben gezeigt, dass die Ursache des Williams-Syndroms in verschiedenen genetischen Veränderungen auf Chromosom 7 liegt. Chromosomen tragen die genetische Information jeder Person und befinden sich im Zellkern des Körpers.
Beim Menschen finden wir 46 Chromosomen, die paarweise verteilt sind. Diese sind von 1 bis 23 nummeriert, mit Ausnahme des letzten Paares aus Geschlechtschromosomen, das bei Frauen als XX und bei Männern als XY bezeichnet wird. Somit kann es in jedem Chromosom eine Unendlichkeit von Genen geben.
Insbesondere ist der beim Williams-Syndrom identifizierte abnormale Prozess eine Mikroselektion oder ein Abbau eines DNA-Moleküls, das dieses Chromosom bestätigt. Normalerweise tritt diese Art von Fehler in der Entwicklungsphase der männlichen oder weiblichen Gameten auf..
Genetische Anomalien finden sich im Bereich 7q11.23, in dem mehr als 25 verschiedene Gene identifiziert wurden, die mit dem charakteristischen klinischen Muster dieser Pathologie zusammenhängen..
Einige der Gene wie Clip2, ELN, GTF21, GTF2IRD1 oder LIMK1 fehlen bei den Betroffenen. Der Verlust von ELN hängt mit Bindegewebe-, Haut- und Herz-Kreislauf-Anomalien zusammen.
Andererseits weisen einige Untersuchungen darauf hin, dass der Verlust der Gene Clip2, GTF2I, GTF2IRD1 und LIMK1 Veränderungen in visuoperzeptiven Prozessen, Verhaltensphänotypen oder kognitiven Defiziten erklären kann.
Darüber hinaus scheint insbesondere das GTF2IRD1-Gen eine herausragende Rolle bei der Entwicklung atypischer Gesichtsmerkmale zu spielen. Das NCF1-Gen scheint seinerseits mit einem hohen Risiko für die Entwicklung von Hypertonie verbunden zu sein.
Bis in die letzten Jahre wurde die Diagnose des Williams-Syndroms ausschließlich anhand der Beobachtung phänotypischer Merkmale (Gesichtsveränderungen, geistige Behinderung, spezifische kognitive Defizite ua) gestellt..
Gegenwärtig wird die Diagnose des Williams-Syndroms jedoch normalerweise in zwei Schritten gestellt: Analyse der klinischen Befunde und bestätigende genetische Studien. Daher umfasst die klinische Diagnose normalerweise:
- Körperliche und neurologische Untersuchung und Bewertung.
- Analyse von Wachstumsparametern.
- Untersuchung des kardiorespiratorischen Systems.
- Nephrourologische Untersuchung.
- Analyse des Kalziumspiegels in Urin und Blut.
- Augenärztliche Analyse.
Andererseits wird eine genetische Analyse verwendet, um das Vorhandensein genetischer Veränderungen zu bestätigen, die mit dem Williams-Syndrom kompatibel sind. Zu den häufigsten Tests gehört die Fluoreszenz-in-situ-Hybridisierungstechnik (FIHS)..
Nach der Extraktion einer Blutprobe wird die In-situ-Hybridisierungstechnik durchgeführt, indem DNA-Sonden markiert werden, die unter fluoreszierendem Licht nachgewiesen werden..
Es gibt keine spezifische Behandlung für das Williams-Syndrom. Diese Pathologie ist jedoch mit mehreren Komplikationen in verschiedenen Organen verbunden, sodass medizinische Interventionen auf die Behandlung dieser Organe ausgerichtet sind.
Die Autoren González Fernández und Uyaguari Quezada betonen, dass alle Interventionen einen ausgeprägten multidisziplinären Charakter haben müssen, um die für dieses Syndrom charakteristische symptomatische Vielfalt behandeln zu können. Darüber hinaus weisen sie je nach betroffenem Bereich auf verschiedene therapeutische Maßnahmen hin:
In diesem Fall erfordern medizinische Komplikationen wie Herzveränderungen oder Fehlbildungen des Bewegungsapparates normalerweise eine Behandlung, die hauptsächlich auf der Verabreichung von Arzneimitteln und chirurgischen Eingriffen beruht. Medizinische Fachkräfte aus verschiedenen Bereichen (Kinderärzte, Kardiologen, Augenärzte usw.) beteiligen sich normalerweise an der Behandlung von körperlichen Symptomen..
Kognitive Defizite wie visuell-perzeptive Veränderungen oder sprachliche Verzögerungen sollten frühzeitig angegangen werden. Kognitive Stimulation und Rehabilitation werden ein entscheidender Faktor für ein autonomes Leben im Erwachsenenalter sein.
Obwohl diejenigen, die vom Williams-Syndrom betroffen sind, normalerweise eine gute soziale Funktion aufweisen, neigen sie in einigen Fällen dazu, übermäßig ängstliche Verhaltensweisen zu zeigen und ausdauernde Verhaltensweisen oder Phobien zu entwickeln.
Daher ist es in diesen Fällen wichtig, einen psychologischen Ansatz durch verschiedene Strategien zu implementieren, die wirksam sind, um diese Probleme oder Schwierigkeiten zu minimieren..

Bisher hat noch niemand einen Kommentar zu diesem Artikel abgegeben.