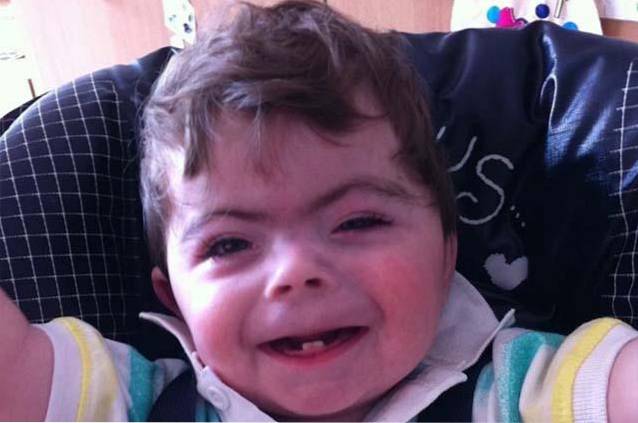
Das Cornelia de Lange-Syndrom ist eine Pathologie genetischen Ursprungs, die durch das Vorhandensein einer signifikanten kognitiven Verzögerung gekennzeichnet ist, die von verschiedenen missbildenden körperlichen Merkmalen begleitet wird.
Auf klinischer Ebene werden drei unterschiedliche klinische Verläufe beobachtet: schwere, mittelschwere und milde. Die Anzeichen und Symptome bestehen normalerweise aus einer atypischen Gesichtskonfiguration, Fehlbildungen des Bewegungsapparates und einer verzögerten kognitiven und psychomotorischen Entwicklung. Darüber hinaus können andere Arten von Anomalien im Zusammenhang mit Herz-, Lungen- und / oder Verdauungsfehlbildungen unterschieden werden..
In Bezug auf den Ursprung des Cornelia de Lange-Syndroms wurde seine Ätiologie unter anderem mit dem Vorhandensein spezifischer Mutationen in den Genen SMC3, SMC1A, NIPBL in Verbindung gebracht. Die Diagnose ist grundsätzlich klinisch und basiert auf physischen und kognitiven Merkmalen. Es wird jedoch normalerweise von einem bestätigenden Gentest begleitet.
Die Behandlung ist auf die Erkennung und Behandlung von medizinischen Komplikationen ausgerichtet. Medizin, Sprachtherapie, neuropsychologische Intervention und Sonderpädagogik sind unerlässlich.
Artikelverzeichnis
Dieses Syndrom wurde ursprünglich 1933 von Dr. Cornelia de Lange beschrieben. Ihre Forschung basierte auf der Untersuchung von zwei Patienten im Alter von 6 und 17 Monaten. Sein Krankheitsbild war durch eine starke Verzögerung des körperlichen Wachstums und der geistigen Entwicklung gekennzeichnet, die mit verschiedenen Missbildungsmerkmalen verbunden war..
Angesichts der Ähnlichkeit beider Fälle ging der erste klinische Bericht über diese Pathologie von einer gemeinsamen und öffentlichen ätiologischen Ursache aus.
Zuvor war es Brachmann (1916) gelungen, einige Autopsiedaten eines Patienten im Kindesalter mit einigen mit dem Cornelia de Lange-Syndrom kompatiblen Merkmalen zu veröffentlichen..
Gegenwärtig wurde das klinische Bild dieses Syndroms in drei verschiedene Phänotypen eingeteilt: schwer, mittelschwer und mild..
Das Cornelia de Lange-Syndrom ist eine seltene genetische Störung angeborener Natur, dh ihre klinischen Merkmale sind von Geburt an offensichtlich. Es handelt sich um eine multisystemische Erkrankung mit Symptomen, die mit einer verzögerten körperlichen und kognitiven Entwicklung, Schädel-Gesichts-Missbildungen oder Fehlbildungen des Bewegungsapparates verbunden sind..
Obwohl der klinische Verlauf und die Schwere dieses Syndroms bei den Betroffenen erheblich variieren können, handelt es sich um eine Krankheit mit einer hohen Sterblichkeitsrate.
Menschen mit Cornelia de Lange-Syndrom zeichnen sich durch eine atypische oder charakteristische Gesichtskonfiguration und eine Verzögerung des prä- und postnatalen Wachstums / der Entwicklung aus..
Am häufigsten treten Lernprobleme, verzögerter Spracherwerb oder Gang- und Verhaltensstörungen auf.
Das Cornelia de Lange-Syndrom ist eine seltene Krankheit in der Allgemeinbevölkerung und wird normalerweise in seltene Krankheiten eingeteilt. Epidemiologische Daten sind nicht genau bekannt. Die Inzidenz wurde auf einen Fall pro 10.000 bis 30.000 Geburten geschätzt..
Bis heute können wir mehr als 400 verschiedene Fälle des Cornelia de Lange-Syndroms finden, die in der medizinischen und experimentellen Literatur beschrieben sind..
Es ist eine Pathologie, die beide Geschlechter in gleicher Anzahl betreffen kann. Einige Autoren wie Gutiérrez Fernández und Pacheco Cumani (2016) schlagen eine leichte Dominanz gegenüber Frauen mit einem Verhältnis von 1,3 / 1 vor.
In Bezug auf den Rest der soziodemografischen Faktoren hat die aktuelle Forschung keine unterschiedliche Prävalenz festgestellt, die mit bestimmten Ländern oder ethnischen und / oder rassischen Gruppen verbunden ist..
Ein großer Teil der diagnostizierten Fälle ist sporadisch, obwohl mehrere betroffene Familien mit einem klaren Muster dominanter Vererbung identifiziert wurden..
Die Anzeichen und Symptome des Cornelia de Lange-Syndroms sind durch ihr breites Beteiligungsmuster gekennzeichnet.
Diese Krankheit ist definiert durch das Vorhandensein charakteristischer Gesichtsmerkmale, Fehlbildungen des Bewegungsapparates in den oberen und unteren Gliedmaßen, eine generalisierte Wachstumsverzögerung vor und nach der Geburt sowie die Entwicklung anderer körperlicher Anomalien..
Als nächstes werden wir einige der häufigsten klinischen Merkmale beim Cornelia de Lange-Syndrom beschreiben:
Bei mehr als 90% der vom Cornelia-Lange-Syndrom Betroffenen ist es möglich, eine Verzögerung der körperlichen Entwicklung oder ein globales Hypogrowth festzustellen. Das Wachstum wird normalerweise sowohl vor als auch nach der Geburt beeinflusst.
Die häufigsten Merkmale bei Neugeborenen sind:
Diese Bedingungen dauern normalerweise bis ins Erwachsenenalter. Darin kann ein Wachstum unterschieden werden, das unter dem für das Geschlecht und das biologische Alter der betroffenen Person erwarteten liegt.
Zusammen mit dieser Art von Veränderungen können einige Anomalien im Zusammenhang mit der Fütterung identifiziert werden. Schwierigkeiten beim Schlucken oder Kauen von Nahrungsmitteln sind in den frühen Lebensphasen häufig.
Die Kombination von Schädel- und Gesichtsveränderungen führt zur Entwicklung eines charakteristischen Gesichtsphänotyps bei Menschen mit Cornelia de Lange-Syndrom..
Einige der häufigsten Anomalien sind:
Die Verzögerung der kognitiven und psychomotorischen Entwicklung ist einer der zentralen klinischen Befunde beim Cornelia-Lange-Syndrom. Ein langsamer Erwerb von Fähigkeiten im Zusammenhang mit motorischer oder geistiger Aktivität wird normalerweise festgestellt.
Die am meisten betroffenen Meilensteine sind das Erlernen des Sitzens, das affektive Lächeln, das Plappern, die unabhängige Bewegung, die Emission der ersten Wörter, das Verständnis und die Befehle, das Füttern, das Gehen oder die unabhängige Toilette.
Bei vielen Betroffenen kann ein durchschnittlicher IQ festgestellt werden, der mit einer leichten oder mittelschweren geistigen Behinderung verbunden ist.
Das Verhalten der vom Cornelia de Lange-Syndrom Betroffenen weist normalerweise einige Besonderheiten auf:
Das Cornelia de Lange-Syndrom ist auch mit der Entwicklung verschiedener medizinischer Komplikationen verbunden.
Die häufigsten Todesursachen oder eine Verschlechterung des medizinischen Zustands der Betroffenen sind:
Die Variabilität der Anzeichen und Symptome des Cornelia de Lange-Syndroms hat eine Klassifizierung seines klinischen Verlaufs ermöglicht:
Es ist normalerweise das ernsteste. Die Veränderungen und Anomalien sind durch das Vorhandensein von körperlichem Unterholz, Fehlbildungen des Bewegungsapparates, abnormalen Gesichtsmerkmalen, Einschränkung der Beweglichkeit der Gelenke, kognitiven Verzögerungen und anderen medizinischen Komplikationen (auditive, okulare, verdauungsfördernde, reno-urologische, kardiale und genitale) gekennzeichnet..
Bei diesem Subtyp sind die physischen Veränderungen normalerweise nicht sehr offensichtlich, insbesondere an den Extremitäten. Die Betroffenen haben normalerweise keine ernsthaften intellektuellen Defizite. Am häufigsten wird die Diagnose über das Neugeborenenstadium hinaus gestellt.
Sein klinischer Verlauf ist grundsätzlich durch klinische Variabilität gekennzeichnet. In den meisten Fällen sind Gesichtsmerkmale vorhanden, aber der Ausdruck der übrigen Anomalien ist variabel..
Der Ursprung des Cornelia-Lange-Syndroms ist mit genetischen Anomalien verbunden. In den untersuchten Fällen konnten spezifische Mutationen in 5 verschiedenen Genen identifiziert werden: NIPBL, SMC1A, HDAC8, RAD21 und SMC3.
Die häufigste Veränderung hängt mit dem NIPBL-Gen zusammen, das bei mehr als der Hälfte der Betroffenen festgestellt wurde. Der Rest der genetischen Anomalien ist weniger häufig.
Alle diese Gene spielen eine herausragende Rolle bei der Produktion von Proteinen im Zusammenhang mit dem Kohäsin-Komplex, die für die Regulation der Chromosomenstruktur und -organisation, die Stabilisierung der genetischen Information in Zellen und die DNA-Reparatur verantwortlich sind..
Darüber hinaus erfüllen sie mehrere grundlegende Funktionen bei der pränatalen Entwicklung der Extremitäten, des Gesichts und anderer Regionen und Körpersysteme.
Die Diagnose des Cornelia de Lange-Syndroms ist klinisch. Derzeit gibt es keinen Labortest, der definitiv auf sein Vorhandensein hinweist. Im medizinischen Bereich werden am häufigsten die von Kline et al..
Diese beziehen sich auf die Identifizierung von kraniofazialen Anomalien in Wachstum und Entwicklung, in den Extremitäten, neurosensorischen und kutanen Veränderungen, Verhaltensstörungen usw..
Darüber hinaus ist es wichtig, eine molekulargenetische Analyse durchzuführen, um das Vorhandensein von Mutationen im Zusammenhang mit dem Cornelia de Lange-Syndrom festzustellen..
Obwohl es keine Heilung für das Cornelia de Lange-Syndrom gibt, beinhaltet sein therapeutischer Ansatz die Gestaltung einer kontinuierlichen medizinischen Nachsorge zusammen mit der Behandlung von Komplikationen..
Die Autoren Gil, Ribate und Ramos (2010) weisen auf einige der am häufigsten verwendeten Ansätze hin.


Bisher hat noch niemand einen Kommentar zu diesem Artikel abgegeben.