Das Prionen Sie sind Proteine ohne Genom oder Nukleinsäuren, die als Infektionserreger wirken. Der Begriff "Prion" bedeutet proteinhaltige infektiöse Partikel (aus dem Englischen proteinhaltige infektiöse Partikel) und wurde vom Neurologen und Nobelpreisträger Stanley B. Prusiner geprägt.
1982 identifizierten Prusiner und seine Kollegen ein infektiöses Proteinpartikel, während sie die Ursachen von Creutzfeldt-Jakob-Erkrankungen (beim Menschen) und der spongiformen Rinderenzephalopathie untersuchten..
Diese seltenen Infektionserreger kommen in der Membran normaler Zellen nur als fehlgefaltete Proteine und / oder mit einer abnormalen dreidimensionalen Struktur vor. Diese Proteine sind für multiple degenerative Erkrankungen und eine sehr hohe Mortalität verantwortlich, die das Nervengewebe und die Struktur des Gehirns beeinflussen..
Sie werden auch Prionkrankheiten genannt. Zu den wichtigsten Betroffenen zählen Kuru, die Gerstmann-Sträussler-Scheinker-Krankheit, das Creutzfeldt-Jakob-Syndrom und tödliche familiäre Schlaflosigkeit..
Artikelverzeichnis
Prionen sind Proteinstrukturen, die in Zellmembranen vorhanden sind. Diese Proteine haben eine veränderte Form oder Konformation [PrP (Sc)].
In Bezug auf seine Vermehrung wird es durch die Umwandlung von Formen erreicht, wie im Fall der Scrapie-Krankheit. Bei dieser Krankheit rekrutieren Prionen PrP (C) (Prionproteine mit unveränderter Konformation), um die Umwandlung in die PrP (Sc) -Isoform zu stimulieren..
Dies erzeugt eine Kettenreaktion, die das infektiöse Material verbreitet und somit die Spülung der Krankheit ermöglicht. Es ist noch nicht bekannt, wie dieser Konvertierungsprozess abläuft.
Diese ungewöhnlichen Proteine, die sich vermehren können, haben keine Nukleinsäuren. Ein Beweis dafür ist, dass sie gegen Röntgenstrahlen und ultraviolette Strahlung beständig sind. Diese Mittel bauen leicht Nukleinsäuren ab.
Prionproteine, aus denen Prionen (PrP) bestehen, kommen im gesamten Körper vor, nicht nur beim Menschen, sondern auch bei anderen gesunden Wirbeltieren. Diese Proteine sind im Allgemeinen resistent gegen Proteasen (Enzyme, die Proteine katalysieren).
Über die Nützlichkeit der Prionproteine PrP (C), der normalen Form des nicht infektiösen Proteins im menschlichen Körper, ist sehr wenig bekannt..
Einigen Forschern ist es jedoch gelungen zu zeigen, dass diese Proteine bei Mäusen die Myelinreparatur in Zellen des peripheren Nervensystems aktivieren. Es wurde auch gezeigt, dass das Fehlen dieser Zellen eine Demyelinisierung solcher Nervenzellen verursacht..
Das Wissen über die Struktur von Prionen beruht hauptsächlich auf den Untersuchungen, die im Bakterium durchgeführt wurden Escherichia coli.
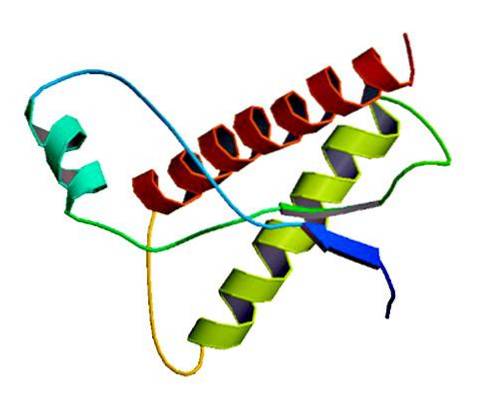
Studien haben gezeigt, dass die Kettenpolypeptide PrP (C) (normal) und PrP (Sc) (infektiös) in der Aminosäurezusammensetzung identisch sind, sich jedoch in ihrer 3D-Konformation und Faltung unterscheiden..
Diese nicht infektiösen Prionen haben beim Menschen 209 Aminosäuren. Sie haben eine Disulfidbindung. Seine Struktur ist alpha-helikal, was bedeutet, dass es spiralförmige Aminosäuren (Alpha-Helices) und wenige flache Aminosäurestränge (Beta-Faltblätter) aufweist..
Dieses Protein kann nicht durch Zentrifugation abgetrennt werden, was bedeutet, dass es nicht sedimentierbar ist. Es wird leicht durch die Breitband-Serinprotease namens Proteinase K verdaut.
Es ist ein infektiöses Protein, das PrP (C) in infektiöse PrP (Sc) -Isoformen mit einer abnormalen Konfiguration oder Form umwandelt.
Über seine 3D-Struktur ist nur sehr wenig bekannt, es ist jedoch bekannt, dass es nur wenige helikale Formen und flachere Stränge oder Beta-Blätter aufweist. Die Verschiebung zur Isoform ist das sogenannte Schlüsselereignis von Prionkrankheiten.
Zelluläre Prionproteine [Prp (C)] befinden sich auf der Zelloberfläche einer Vielzahl von Organen und Geweben. Über die physiologischen Funktionen von Prionen im Körper ist sehr wenig bekannt. Trotzdem zeigen Experimente an Mäusen mögliche Funktionen wie:
Es wurde gezeigt, dass PrP (C) mit Glutamatrezeptoren (ionotrop und metabotrop) wirkt. PrP (C) ist als Rezeptor für synaptotoxische Oligomere des Zelloberflächenpeptids Aβ beteiligt.
Bei Mäusen der Murinae-Familie wurde entdeckt, dass die Prionproteine PrP (C) innerhalb weniger Tage nach der Implantation in der Embryonalentwicklung exprimiert werden.
Dies weist darauf hin, dass sie bei der Entwicklung dieser kleinen Säugetiere eine Rolle spielen. Rolle, die laut den Forschern mit der Regulation der Neuritogenese zusammenhängt (Produktion von Axonen und Dendriten von Neuronen).
Sie wirken auch auf das axonale Wachstum. Diese Prionproteine sind sogar an der Entwicklung des Kleinhirnkreislaufs beteiligt. Aus diesem Grund wird angenommen, dass das Fehlen dieser PrP (C) -Prionen eine Verzögerung in der motorischen Entwicklung von Nagetieren mit sich bringt..
In Studien zur Überexpression von PrP (C) durch Genorientierung wurde festgestellt, dass das Fehlen dieser Prionen Probleme mit der Blutversorgung einiger Teile des Gehirns verursacht (akute zerebrale Ischämie)..
Dies bedeutet, dass Prionproteine als Neuroprotektoren fungieren. Zusätzlich wurde gezeigt, dass eine Überexpression von PrP (C) Verletzungen durch Ischämie reduzieren oder verbessern kann..
Die physiologische Rolle von Prp (C) bei der Aufrechterhaltung des peripheren Myelins wurde kürzlich entdeckt.
Während einer Laborstudie wurde festgestellt, dass Labormäuse in Abwesenheit des Prionproteins Defizite in den Nerven entwickelten, die Informationen aus dem Gehirn und dem Rückenmark transportieren, was als periphere Neuropathie bezeichnet wird.
Es gibt einige Proteine, die Prionen ähnlich sind, und diese befinden sich in anderen Teilen des Körpers als im Gehirn.
Die Funktion solcher Proteine besteht darin, den Zelltod zu initiieren, zu regulieren und / oder zu kontrollieren, wenn der Organismus angegriffen wird (zum Beispiel durch Vironen), wodurch die Ausbreitung des Pathogens verhindert wird..
Diese besondere Funktion dieser Proteine lässt die Forscher über die mögliche Bedeutung nichtinfektiöser Prionen im Kampf gegen Krankheitserreger nachdenken..
Eine am Stowers Institute in Missouri, USA, durchgeführte Studie zeigte, dass PrP-Prionen eine Rolle bei der Aufrechterhaltung des Langzeitgedächtnisses spielen können.
Die Studie ergab, dass bestimmte Prionproteine so gesteuert werden können, dass sie die physiologischen Funktionen des Langzeitgedächtnisses aufrechterhalten..
Eine Untersuchung an Prionproteinen, die in Stammzellen des Blutgewebes exprimiert werden, ergab, dass alle diese Stammzellen (hämatopoetisch) Prionproteine in ihrer Zellmembran exprimieren. Für das, was man glaubt, dass sie am komplexen und sehr wichtigen Prozess der Zellerneuerung beteiligt sind.
Pathologien prionischen Ursprungs werden als fortschreitende degenerative Hirnstörungen erkannt. Sie können Rinder, Hirsche, Karibu, Schafe und sogar Menschen angreifen.
Diese Krankheiten werden durch eine Veränderung der Struktur der PrP (C) -Proteine verursacht, deren spezifische Funktionen bis heute ungewiss sind. Prionpathologien können ohne bekannte Ursache auftreten. Sie können einen vererbten genetischen Ursprung haben und auch infektiös-ansteckend übertragen werden.
Prionen verursachen familiäre, sporadische und ansteckende Krankheiten. Familiäre Prionkrankheiten sind solche, die vererbbar sind. Sporadische Pathologien sind am häufigsten und treten ohne bekannte Ursachen auf..
Ansteckende Krankheiten gelten als selten, sie werden von Person zu Person, von Tier zu Tier, von Person zu Tier und umgekehrt übertragen. Die Ursachen sind vielfältig und reichen vom Verzehr von kontaminiertem Fleisch, Kannibalismus, Transfusionen bis zur Manipulation kontaminierter chirurgischer Geräte.
Die häufigsten Prionkrankheiten sind:
Es gilt als die häufigste Prionkrankheit unter Menschen und ist eine kosmopolitische Krankheit, dh sie ist weltweit verbreitet. Es kann erblich (familiär), sporadisch oder ansteckend sein.
Patienten mit Symptomen wie Demenz, Rucken oder plötzlichen unwillkürlichen Bewegungen sowie Defiziten des Zentralnervensystems.
Abhängig von der Behandlung und Form der Krankheit kann der Tod zwischen 4 Monaten und 2 Jahren nach dem Erwerb der Krankheit eintreten. Die Diagnose ist schwierig, sie wird normalerweise gestellt post morten, während der Autopsie.
Es ist eine Krankheit, die durch Prionen in einem vererbbaren oder autosomal dominanten infektiösen Gehirnprozess verursacht wird. Die Krankheit manifestiert sich bei Menschen im Alter von 40 bis 60 Jahren.
Diese Menschen haben Probleme, Wörter (Dysarthrie), Rucke oder plötzliche unwillkürliche Bewegungen zu artikulieren, wobei Aggressivität häufig ist.
Sie zeigen eine Kleinhirnentartung, die von einem unsteten Gang begleitet wird. Es ist auch möglich, unter anderem Hyporeflexie, Taubheit, Blicklähmung, Demenz zu beobachten. Die Lebenserwartung beträgt ca. 5 Jahre oder etwas länger.
Es ist eine sehr seltene Krankheit, bis zu einem Punkt, an dem 2 bis 3 Fälle pro 100 Millionen Einwohner auftreten. Die Pathologie ähnelt der Gerstmann-Sträussler-Scheinker-Krankheit.
Die klinischen Manifestationen des Proteins weisen auf eine geringe Resistenz gegen Proteasen hin, einige sind mehr und andere weniger empfindlich gegenüber diesen Enzymen.
Die Symptome, die Patienten zeigen, sind: Probleme mit Sprache und kognitiven Beeinträchtigungen, Verlust von Neuronen in dem Bereich, in dem das Gehirn Bewegungen steuert und Muskelkoordination durchführt.
Die Krankheit tritt häufig bei älteren Patienten (70 Jahre) auf und die geschätzte Lebensdauer nach einer Infektion beträgt ungefähr 20 Monate.
Es ist eine erbliche oder familiäre Erkrankung, die auch sporadisch auftreten kann. Es ist bekannt, dass die Krankheit auf eine erbliche oder autosomal dominante Mutation zurückzuführen ist.
Die Patienten zeigen Symptome wie kumulative Schlaf- und Schlafstörungen, Demenz, kognitive Beeinträchtigungen, sogar Probleme mit Bluthochdruck, Tachykardie, Hyperhidrose und andere..
Das betroffene Alter ist ziemlich breit und liegt zwischen 23 und 73 Jahren, das Durchschnittsalter beträgt jedoch 40 Jahre. Die einmal infizierte Lebensdauer beträgt etwas mehr als 6 Jahre.
Diese Prionkrankheit wurde nur bei den Einwohnern von Papua-Neuguinea festgestellt. Es ist eine Krankheit, die mit Kannibalismus und der kulturellen Tradition des Trauerritus zusammenhängt, bei dem diese Menschen Gehirn oder menschliches Fleisch essen.
Menschen, die die Krankheit tragen, haben normalerweise unkontrollierbare und unwillkürliche Bewegungen in verschiedenen Körperteilen.
Sie zeigen Zittern, Verlust der Kontrolle über Bewegungen und Verlust der Muskelkoordination. Die Lebenserwartung bei infizierten Menschen beträgt zwei Jahre.
Zu den durch Prionen bei Tieren hervorgerufenen Pathologien gehört die spongiforme Rinderenzephalopathie. Diese Krankheit verursachte in Europa, in der öffentlichen Gesundheit, bei Tieren und in der Wirtschaft der betroffenen Länder Chaos.
Andere Krankheiten bei Tieren sind Scrapie, übertragbare Nerz-Enzephalopathie, chronische Abfallkrankheit (bei Hirschen) und spongiforme Katzen-Enzephalopathie..
Diese Krankheiten, wie sie auch beim Menschen auftreten, sind nicht wirksam zu behandeln. Daher ist eine Vorbeugung unerlässlich, insbesondere nach Infektionen beim Menschen, die durch den Verzehr von Fleisch von infizierten Kühen verursacht wurden..
Bisher ist kein Heilmittel für Prionkrankheiten bekannt. Die Behandlung ist symptomatisch. Den Patienten wird empfohlen, Palliativversorgung und Gentests zu planen, und es wird empfohlen, Familienmitglieder zu beraten.
Eine Vielzahl von Medikamenten wurde bei Patienten mit Prionkrankheiten wie Virostatika, Antitumoren, Medikamenten gegen Krankheiten wie Parkinson, Behandlungen zur Immunsuppression, Antibiotika, Antimykotika und sogar Antidepressiva getestet..
Derzeit gibt es jedoch keine Hinweise darauf, dass einige davon die Symptome lindern oder das Überleben der Patienten verbessern..
Prionen sind gegen eine Vielzahl physikalischer und chemischer Veränderungen resistent. Es werden jedoch verschiedene Techniken verwendet, um eine Kontamination von Patienten mit kontaminierten chirurgischen Instrumenten zu vermeiden..
Zu den am häufigsten verwendeten Techniken gehört das Sterilisieren der Ausrüstung in einem Autoklaven bei 132 ° C für eine Stunde und das anschließende Eintauchen der Instrumente in Natriumhydroxid für mindestens eine weitere Stunde..
Andererseits hat die Weltgesundheitsorganisation (WHO) Maßnahmen entwickelt, um die Ausbreitung von Prionkrankheiten zu verhindern. Diese Organisation legt Normen für den Umgang mit verbotenen oder potenziell riskanten Geweben wie Augen, Gehirn, Darm, Mandeln und Rückenmark fest.



Bisher hat noch niemand einen Kommentar zu diesem Artikel abgegeben.