
Das Prader-Willi-Syndrom ((SPW) ist eine multisystemische Pathologie, die einen angeborenen genetischen Ursprung hat. Es ist eine komplexe Krankheit, die Appetit, Wachstum, Stoffwechsel, Verhalten und / oder kognitive Funktion beeinflusst.
Auf klinischer Ebene ist diese Krankheit im Kindesalter durch das Vorhandensein verschiedener medizinischer Befunde wie Muskelschwäche, Essstörungen oder allgemeine Entwicklungsverzögerungen gekennzeichnet..
Darüber hinaus weist ein großer Teil der vom Prader-Willi-Syndrom betroffenen Personen auf kognitiver und verhaltensbezogener Ebene eine moderate geistige Beeinträchtigung oder Verzögerung auf, die mit verschiedenen Lern- und Verhaltensproblemen einhergeht.
Obwohl das Prader-Willi-Syndrom als seltene oder seltene Krankheit angesehen wird, weisen zahlreiche Studien darauf hin, dass es eine der häufigsten Pathologien im genetischen Bereich ist. Die Diagnose dieser Krankheit wird hauptsächlich auf der Grundlage klinischer Befunde und komplementärer Gentests gestellt..
In Bezug auf die Behandlung wurde noch keine Heilung für das Prader-Willi-Syndrom identifiziert. Daher ist der therapeutische Ansatz auf die Behandlung der Symptome und Komplikationen ausgerichtet, wobei Fettleibigkeit der medizinische Befund ist, der die größte Bedrohung für die Betroffenen darstellt.
In Bezug auf die Prognose und die Lebensqualität hängen beide von der Schwere der damit verbundenen medizinischen Probleme und den möglicherweise auftretenden Verhaltens- oder kognitiven Störungen ab.
Artikelverzeichnis
Verschiedene klinische Berichte weisen darauf hin, dass das Prader-Willi-Syndrom (PWS) erstmals 1887 von J. L. Down beschrieben wurde, nachdem bei einem seiner Patienten eine „Polysarkia“ diagnostiziert worden war..
Es waren jedoch Dr. Prader, Labhart und Willi, die 1956 weitere 9 Fälle beschrieben und dieser Pathologie ihren Namen gaben. Darüber hinaus wurden die Merkmale und diagnostischen Kriterien des Prader-Willi-Syndroms von Holm et al..
Das Prader-Willi-Syndrom ist eine angeborene genetische Veränderung, dh es handelt sich um eine Pathologie, die vom Moment der Geburt an vorliegt und das Individuum während seines gesamten Lebens betrifft, wenn keine kurative therapeutische Intervention erfolgt..
Diese Pathologie stellt einen komplexen klinischen Verlauf dar, der durch zahlreiche medizinische Manifestationen gekennzeichnet ist.
Obwohl der Phänotyp des Prader-Willi-Syndroms heute genauer bekannt ist, wurden in den letzten 25 Jahren erhebliche Fortschritte bei der Analyse und dem Verständnis dieser Krankheit erzielt.
Die Expression des Prader-Willis-Syndroms ist vielfältig und betrifft tendenziell mehrere Systeme und Strukturen, wobei die meisten Veränderungen mit einer hypothalamischen Dysfunktion zusammenhängen.
Der Hypothalamus ist eine neurologische Struktur, die eine wesentliche Rolle bei der Steuerung homöostatischer Funktionen spielt: die Regulierung von Hunger, Durst, Schlaf-Wach-Zyklen oder die Regulierung der Körpertemperatur.
Darüber hinaus setzt der Hypothalamus verschiedene Hormone an verschiedene Drüsen frei: Wachstum, Sexualität, Schilddrüse usw..
Schließlich müssen wir darauf hinweisen, dass das Prader-Willis-Syndrom auch in der medizinischen und experimentellen Literatur mit anderen Begriffen wie dem Prader-Labhart-Willi-Syndrom oder mit dem Akronym PWS referenziert erscheinen kann..
Andere Synonyme sind das Labhart-Willi-Syndrom, das Praser-Labhart-Willi-Fancone-Syndrom oder das Hypogenital-Dystrophie-Syndrom..
Das Prader-Willi-Syndrom (PWS) ist eine seltene genetische Erkrankung. Der Begriff seltene Krankheit (ER) wird verwendet, um jene Pathologien zu bezeichnen, die selten sind oder nur wenige Menschen, die darunter leiden.
Derzeit wird geschätzt, dass das Prader-Willi-Syndrom eine Pathologie mit einer ungefähren Häufigkeit von 1 Fall pro 10.000 bis 30.000 Menschen weltweit ist.
Andererseits wurde in Bezug auf die Verteilung nach Geschlecht beobachtet, dass diese Pathologie Männer und Frauen gleichermaßen betrifft und nicht mit ethnischen Gruppen oder geografischen Regionen assoziiert ist..
Darüber hinaus gilt das Prader-Willi-Syndrom als Hauptursache für Fettleibigkeit genetischen Ursprungs.
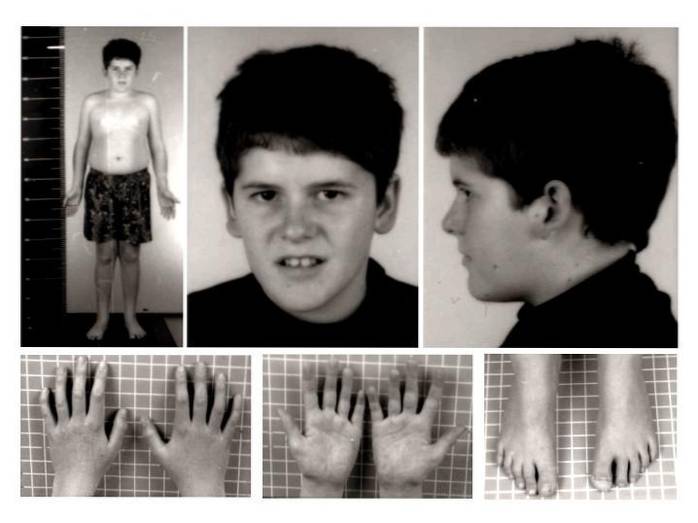
Auf klinischer Ebene wurde das Prader-Willi-Syndrom traditionell mit neonataler Hypotonie, Hypogonadismus, Hyperphagie, Fettleibigkeit, Kleinwuchs, allgemeiner Entwicklungsverzögerung, mäßiger geistiger Behinderung, atypischem Gesichtsaussehen und verschiedenen Verhaltensänderungen in Verbindung gebracht..
Trotzdem ist die klinische Expression dieser Pathologie sehr heterogen und variiert signifikant zwischen den betroffenen Personen..
Darüber hinaus variieren die charakteristischen Anzeichen und Symptome des Prader-Willi-Syndroms in der Regel mit der biologischen Entwicklung, sodass wir unterschiedliche klinische Befunde in der fetalen und neonatalen Phase, in der Kindheit oder frühen Kindheit, im Schulstadium und schließlich im Stadium Teen beobachten können.
José A. del Barrio del Campo und Mitarbeiter beschreiben systematisch die charakteristischsten Veränderungen im biomedizinischen, psychomotorischen, kognitiven und Verhaltensbereich:
Die charakteristischsten körperlichen Anzeichen und Symptome sind Störungen wie; Hypotonie, Fehlbildungen oder Deformitäten des Bewegungsapparates, reduziertes oder geringes Gewicht und geringe Körpergröße, übermäßiger Appetit, Fettleibigkeit, Hypogonadismus, Schlafstörungen, Atemwegserkrankungen, atypische leichte Merkmale, Veränderung der Regulierung der Körpertemperatur ua.
Vorhandensein oder Entwicklung eines reduzierten Muskeltonus. Die Muskelschwäche bei dieser Pathologie ist im Nacken und Rumpf besonders ausgeprägt, insbesondere im Neugeborenenstadium und in den ersten Lebensmonaten. Mit der biologischen Entwicklung neigt der Muskeltonus daher dazu, sich zu verbessern.
In diesem Fall ist es üblich, die Entwicklung von Skoliose oder Abweichung der Wirbelsäule, eine schlechte Ausrichtung der unteren Gliedmaßen (Genu Valgus) oder das Vorhandensein von Plattfüßen zu beobachten..
Darüber hinaus können auch andere Arten von angeborenen Anomalien beobachtet werden, wie z. B. eine Verringerung der Größe der Füße und Hände, Hüftdysplasie und das Vorhandensein von sechs Fingern..
Insbesondere zum Zeitpunkt der Geburt ist sowohl die Größe als auch das Gewicht des betroffenen Kindes aufgrund seiner Entwicklung und seines Geschlechts geringer als erwartet. Trotz der Tatsache, dass Standardwerte im Erwachsenenalter erreicht werden können, verändert die langsame Wachstumsrate tendenziell die Werte für Größe und Gewicht bei Erwachsenen.
Es ist üblich, bei Menschen mit Prader-Willi-Syndrom einen unersättlichen Appetit zu beobachten, der durch eine Besessenheit oder Fixierung auf Nahrung gekennzeichnet ist. Aufgrund der Aufnahme großer Mengen an Nahrungsmitteln neigen die Betroffenen dazu, Fettleibigkeit und andere damit verbundene medizinische Komplikationen wie Typ-II-Diabetes mellitus zu entwickeln.
Das Vorhandensein von Genitalveränderungen ist ebenfalls häufig. Insbesondere Hypogonadismus oder teilweise Entwicklung der äußeren Genitalien sind sehr häufig. In den meisten Fällen erreicht die Pubertätsentwicklung nicht das Endstadium oder das Erwachsenenstadium.
Schnarchen, erhöhte Häufigkeit oder Atemstillstand treten häufig während der Schlafphasen auf. Daher neigen die Betroffenen dazu, verschiedene Veränderungen im Zusammenhang mit Fragmentierung, Schlafverzögerung oder dem Vorhandensein periodischer Erwachungen zu zeigen.
Anomalien und Missbildungen des Bewegungsapparates können auch die kraniofazialen Merkmale beeinträchtigen. Es ist möglich, einen schmalen Schädel, Augenstrabismus, schlecht pigmentierte Haut und Haare, einen kleinen Mund und dünne Lippen, Zahnfehlbildungen usw. zu beobachten..
Menschen, die vom Prader-Willi-Syndrom betroffen sind, haben normalerweise Probleme im Zusammenhang mit der Regulierung der Körpertemperatur, und ein weiterer wichtiger Befund ist die hohe Schmerzresistenz.
Aufgrund des Vorhandenseins von Fehlbildungen des Bewegungsapparates und eines verringerten Muskeltonus ist die psychomotorische Entwicklung langsamer und betrifft alle Bereiche.
Die Betroffenen haben normalerweise Serienschwierigkeiten bei der Durchführung von Aktivitäten, die eine oder mehrere motorische Ausführungen erfordern.
In Bezug auf kognitive Einschränkungen haben die meisten Betroffenen eine leichte oder mittelschwere geistige Behinderung.
Darüber hinaus weisen sie in der Regel einige Bereiche auf, die stärker betroffen sind, z. B. die sequentielle Verarbeitung von Informationen, das aktuelle oder Kurzzeitgedächtnis, die Lösung von Rechenproblemen, die auditive Verarbeitung verbaler Informationen, die Veränderung der Aufmerksamkeit und Konzentration sowie das Vorhandensein kognitiver Starrheit.
Andererseits ist die Sprache ein weiterer Bereich, der bei Personen mit Prader-Willi-Syndrom erheblich betroffen ist. In der Regel werden unter anderem Verzögerungen beim Erwerb phonologischer Fähigkeiten, ein schlechter Wortschatz und eine Änderung der grammatikalischen Konstruktion beobachtet..
Verhaltensprobleme und -veränderungen sind weitere typische Befunde, die beim Prader-Willi-Syndrom beobachtet werden können. Sie variieren normalerweise je nach Alter oder Reifungsstadium, in dem sich die betroffene Person befindet. Einige der häufigsten Verhaltensmerkmale sind jedoch:
Verschiedene aktuelle Untersuchungen haben gezeigt, dass Verhaltensänderungen mit zunehmendem Alter tendenziell zunehmen und sich daher tendenziell verschlechtern, was sich allgemein auf soziale, familiäre und emotionale Bereiche auswirkt.
Wie wir in mehreren Abschnitten oben ausgeführt haben, hat das Prader-Willi-Syndrom einen genetischen Ursprung.
Obwohl derzeit große Kontroversen über die spezifischen Gene bestehen, die für diese Pathologie verantwortlich sind, zeigen alle Daten, dass sich die ätiologische Veränderung auf Chromosom 15 befindet.
Während der genetischen Untersuchung dieser Pathologie gab es mehrere Beiträge. Burtler und Palmer (1838) entdeckten das Vorhandensein von Anomalien im langen Arm von Chromosom 15 des väterlichen Elternteils, während Nicholls (1989) beobachtete, dass die Störung in anderen Fällen mit chromosomalen Veränderungen der Mutter zusammenhängt (Rosell-Raga, 2003)..
Abgesehen davon ist die am meisten akzeptierte Theorie über den Ursprung dieser Pathologie der Verlust oder die Inaktivierung verschiedener Gene der väterlichen Expression, die sich in der 15q11-13-Region von Chromosom 15 befinden.
Die Diagnose des Prader-Willi-Syndroms besteht aus zwei Grundkomponenten: der Analyse klinischer Befunde und Gentests..
In Bezug auf die Erkennung von Anzeichen und Symptomen von Indikatoren sowohl bei Babys als auch bei älteren Kindern ist es wichtig, eine detaillierte, individuelle und familiäre Krankengeschichte zu erstellen. Ebenso ist es wichtig, eine körperliche und neurologische Untersuchung durchzuführen..
Wenn aufgrund dieser Verfahren ein diagnostischer Verdacht besteht, müssen verschiedene ergänzende Tests verschrieben werden, um das Vorhandensein genetischer Veränderungen und Anomalien festzustellen..
Insbesondere werden rund 90% der Fälle durch DNA-Methylierungstests und andere zusätzliche Tests definitiv diagnostiziert..
Darüber hinaus ist es auch möglich, eine pränatale Diagnose dieser Erkrankung zu stellen, hauptsächlich in Familien mit einer Vorgeschichte des Prader-Willi-Syndroms..
Insbesondere ermöglicht der Amniozentese-Test die Extraktion von Embryo-Proben für die Durchführung der entsprechenden Gentests.
Derzeit gibt es keine Heilung für das Prader-Willi-Syndrom. Wie bei anderen seltenen Krankheiten beschränken sich die Behandlungen auf die Symptomkontrolle und die Verbesserung der Lebensqualität der Betroffenen.
Einer der grundlegenden Aspekte wird jedoch die Ernährungs- und Ernährungskontrolle sein, da Fettleibigkeit die Hauptursache für Morbidität und Mortalität in dieser Pathologie ist..
Andererseits erfordert das Vorhandensein kognitiver und Verhaltensänderungen die Intervention spezialisierter Fachkräfte sowohl bei der kognitiven Rehabilitation als auch bei der Behandlung von Verhaltensstörungen.


Bisher hat noch niemand einen Kommentar zu diesem Artikel abgegeben.