
Das Pallister-Killian-Syndrom, Auch als Tetrasomie 12 bekannt, handelt es sich um eine seltene Krankheit genetischen Ursprungs, die durch ein breites Spektrum der Beteiligung mehrerer Organe gekennzeichnet ist.
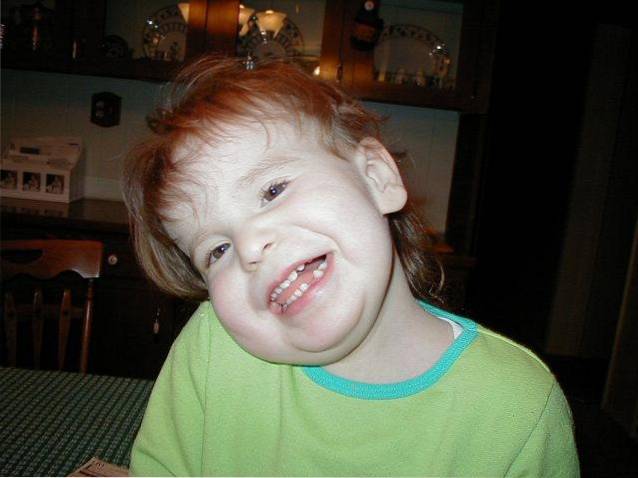
Klinisch ist diese Pathologie durch geistige Behinderung, psychomotorische Retardierung, Muskelhypotonie, einen atypischen Gesichtsphänotyp, Pigmentanomalien in der Haut und Alopezie definiert. Darüber hinaus können auch andere Arten von medizinischen Komplikationen im Zusammenhang mit Missbildungen in verschiedenen Körpersystemen oder Anfällen auftreten..
Der ätiologische Ursprung dieser Krankheit ist mit einer im Mosaik verteilten genetischen Störung verbunden. Insbesondere ist es auf das Vorhandensein eines zusätzlichen Chromosoms 12 in einigen Körperzellen zurückzuführen.
Die Diagnose des Pallister-Killiam-Syndroms kann sowohl vor als auch nach der Geburt gestellt werden. Das Hauptziel ist die Identifizierung der klinischen Merkmale und die Verwendung einer bestätigenden genetischen Studie..
Dieses Syndrom hat eine hohe Sterblichkeitsrate. Der pharmakologische medizinische Ansatz und die rehabilitative Behandlung können jedoch wichtige Vorteile für die Lebensqualität und den klinischen Status der Betroffenen bieten.
Artikelverzeichnis
Diese Krankheit wurde ursprünglich von Pallister im Jahr 1977 beschrieben. In den ersten Veröffentlichungen berichtete dieser Forscher über zwei Fälle von erwachsenen Patienten, deren Verlauf durch verschiedene Befunde gekennzeichnet war: Anfälle, Muskelhypotonie, intellektuelles Defizit, muskuloskelettale und organische Missbildungen, rauhe Gesichts- und Hautfarbe Änderungen.
Parallel dazu beschrieben Teschler-Nicola und Killiam 1981 dasselbe Krankheitsbild bei einem dreijährigen Mädchen.
Daher wurde in den ersten klinischen Berichten allgemein auf einen medizinischen Zustand Bezug genommen, der durch die Kombination von Anfällen, geistiger Behinderung und einem charakteristischen physischen Phänotyp gekennzeichnet ist..
Darüber hinaus konnte Gilgenkratz bereits 1985 im ersten Fall während der Schwangerschaftsphase identifizieren, was heute dank moderner Diagnosetechniken üblich ist.
Das Pallister-Killiam-Syndrom ist eine Art genetischer Mosaikkrankheit. In diesem Fall betrifft die Chromosomenveränderung nur einige Körperzellen. Eine breite Beteiligung verschiedener Körpersysteme und Organismen wird identifiziert.
Es ist unter anderem gekennzeichnet durch geistige Behinderung, Muskelhypotonie, die Entwicklung charakteristischer Gesichtsmerkmale, Veränderung der Hautpigmentierung oder Haarwuchs..
Darüber hinaus ist das Pallister-Kiliam-Syndrom eine seltene Krankheit angeborenen Ursprungs, die in der medizinischen Literatur eine Vielzahl von Namen erhalten kann:
Die Prävalenzzahlen für das Pallister-Killiam-Syndrom sind nicht genau bekannt. Es wurden nicht viele endgültige Diagnosen gestellt und die meisten davon wurden nicht in der medizinischen Literatur veröffentlicht.
Daher definieren alle Autoren und Institutionen dieses Syndrom als eine seltene oder seltene genetische Pathologie in der Allgemeinbevölkerung..
Vor ungefähr 15 Jahren wurde das Pallister-Killiam-Syndrom in nur ungefähr 100 Fällen weltweit identifiziert. Derzeit sind mehr als 200 Betroffene betroffen.
Epidemiologische Untersuchungen haben die Inzidenz dieser Krankheit auf etwa 5,1 Fälle pro Million neugeborener Kinder geschätzt, obwohl Autoren wie Toledo-Bravo de la Laguna und Mitarbeiter sie auf 1 / 25.000 schätzen.
Eine höhere Prävalenz im Zusammenhang mit den soziodemografischen Merkmalen der Betroffenen wurde nicht festgestellt. Das Pallister-Killian-Syndrom kann in jedem Geschlecht, jeder technischen und / oder rassischen Gruppe auftreten.
Im klinischen Verlauf des Pallister-Killian-Syndroms kann eine Vielzahl von Anzeichen und Symptomen festgestellt werden. Alle von ihnen sind mit kraniofazialen und / oder muskuloskelettalen Anomalien und kognitiven Veränderungen verbunden.
Die Entwicklung von Schädel-Gesichts-Missbildungen von der Schwangerschaftsphase bis zum postnatalen und kindlichen Wachstum ist eines der charakteristischsten medizinischen Anzeichen des Pallister-Killiam-Syndroms..
Die häufigsten Anzeichen und Symptome sind Anomalien in den verschiedenen Schädel- und Gesichtsstrukturen, die zu einem rauen und atypischen Erscheinungsbild führen:
Obwohl es weniger signifikant als Gesichtsveränderungen ist, ist es sehr häufig, dass bei Patienten, die vom Pallister-Syndrom betroffen sind, mehrere Anomalien des Bewegungsapparates beobachtet werden:
Abnormalitäten im Zusammenhang mit Muskelstruktur und Mobilität sind weitere klinische Merkmale des Pallister-Killian-Syndroms:
Muskelhypotonie bezieht sich auf die Identifizierung von abnormal reduziertem Muskeltonus oder Muskelverspannungen. Auf visueller Ebene können Schlaffheit und Labilität in verschiedenen Muskelgruppen beobachtet werden, insbesondere in den Extremitäten..
Daher führt die Muskel- und Skelettpathologie zu einer signifikanten Verzögerung beim Erwerb unterschiedlicher motorischer Fähigkeiten, sowohl in der Neugeborenen- als auch in der Kindheitsperiode..
Obwohl die Entwicklungsperioden unter den Betroffenen unterschiedlich sind, enthält der häufigste Kalender die folgenden Meilensteine:
Ein weiterer stark betroffener Bereich ist das Nervensystem. In den meisten Fällen hängen die Anzeichen und Symptome hauptsächlich mit Anfällen und geistiger Behinderung zusammen:
Obwohl sie weniger häufig sind, können auch andere Arten von medizinischen Komplikationen auftreten:
Der Ursprung des Pallister-Killian-Syndroms ist mit einer genetischen Mosaikanomalie auf Chromosom 12 verbunden. Sie betrifft nur das genetische Material einiger Zellen im Körper..
Chromosomen sind Teil des Zellkerns aller im menschlichen Körper vorkommenden Zellen. Sie bestehen aus einer Vielzahl biochemischer Komponenten und enthalten die genetische Information jedes Einzelnen..
Menschen haben 46 verschiedene Chromosomen, die paarweise organisiert und von 1 bis 23 nummeriert sind. Außerdem hat jedes Chromosom auf individueller Ebene einen kurzen Bereich oder Arm mit der Bezeichnung "p" und einen langen Bereich mit der Bezeichnung "q"..
Die Abnormalität betrifft Chromosom 12 und führt zum Vorhandensein eines Chromosoms mit einer abnormalen Struktur, das als Isochromosom bezeichnet wird.
Daher neigt dieses Chromosom dazu, zwei kurze Arme anstelle eines von jeder p (kurz) und langen (q) Konfiguration zu haben..
Infolgedessen verändert das Vorhandensein von zusätzlichem und / oder abnormalem genetischem Material den normalen und effizienten Verlauf der körperlichen und kognitiven Entwicklung der betroffenen Person, was zu den klinischen Merkmalen des Pallister-Killian-Syndroms führt..
Das Pallister-Killian-Syndrom kann während der Schwangerschaft oder im postnatalen Stadium anhand der klinischen Merkmale und der Ergebnisse verschiedener Labortests identifiziert werden..
Während der Schwangerschaft werden am häufigsten Ultraschalluntersuchungen, Amniozentese oder Chorionzottenproben durchgeführt. In diesem Sinne kann uns die Analyse des genetischen Materials des Embryos eine Bestätigung dieser Pathologie durch die Identifizierung kompatibler Anomalien bieten..
Wenn die Diagnose jedoch nach der Geburt gestellt wird, ist Folgendes unerlässlich:
Es wurden keine spezifischen Therapien zur Behandlung von Menschen mit Pallister-Killian-Syndrom entwickelt..
Dieses Syndrom ist normalerweise mit einer schlechten neurologischen Prognose und hohen Sterblichkeitsraten verbunden. Rehabilitationsbehandlung, Sonderpädagogik und Ergotherapie können jedoch eine gute funktionelle Prognose und eine Verbesserung der Lebensqualität der Betroffenen bieten..
Zum Beispiel beschreiben Méndez und sein Arbeitsteam (2013) einen Fall von Rehabilitationsbehandlung, der gekennzeichnet ist durch:


Bisher hat noch niemand einen Kommentar zu diesem Artikel abgegeben.