
Das DiGeorge-Syndrom ist eine Pathologie genetischen Ursprungs, die sich in der Entwicklung von Missbildungen im Zusammenhang mit der Struktur von Herz, Gesicht, Thymus und Nebenschilddrüsen manifestiert.
Auf klinischer Ebene werden sie eine Vielzahl von medizinischen Komplikationen hervorrufen, darunter Immunschwäche, Hypokalzämie, Herzerkrankungen und psychiatrische Störungen..

In Bezug auf den ätiologischen Ursprung ist es mit einer genetischen Veränderung von Chromosom 22 verbunden. Aus diesem Grund wird es auch als 22q11.2-Deletionssyndrom bezeichnet..
Die Diagnose basiert auf der Identifizierung klinischer Kardinalzeichen durch körperliche Untersuchung und verschiedene Labortests: analytische und immunologische Untersuchung, Ultraschall des Abdomens, Echokardiogramme und genetische Untersuchungen, die im Wesentlichen auf fluoreszierender In-situ-Hybridisierung (FISH) beruhen..
Schließlich konzentriert sich die Behandlung dieser Pathologie auf die Korrektur organischer Missbildungen und die Kontrolle medizinischer Komplikationen. Daher werden üblicherweise T-Lymphozyten-Therapie, Kalziumpräparate, Korrekturoperationen usw. verwendet..
Artikelverzeichnis
Diese Pathologie wurde ursprünglich 1965 vom amerikanischen Kinderarzt Angelo M. DiGeorge beschrieben. In seinem klinischen Bericht beschrieb DiGeroge eine angeborene Pathologie, die durch die mangelnde Entwicklung oder Abwesenheit der Nebenschilddrüse und des Thymus definiert ist.
Anschließend beschrieb Chapelle 1918 speziell die angeborenen Defekte, die sich aus dieser Pathologie ergeben. Daher wurde das DiGeorge-Syndrom als zweite Ursache für angeborene Herzfehler nach dem Down-Syndrom bezeichnet.
Schließlich wurde diese Pathologie klinisch durch die klassische Triade von Immunschwäche, Endokrinopathie mit Hypokalzämie und Herzerkrankungen charakterisiert..
Darüber hinaus impliziert die breite symptomatische Heterogenität der auf Chromosom 22 befindlichen Deletionen in vielen Fällen die Unterscheidung von drei verschiedenen Arten von Pathologien auf klinischer Ebene:
- DiGeorge-Syndrom
- Velocardiofacial Syndrom
- Kardiofaziales Syndrom
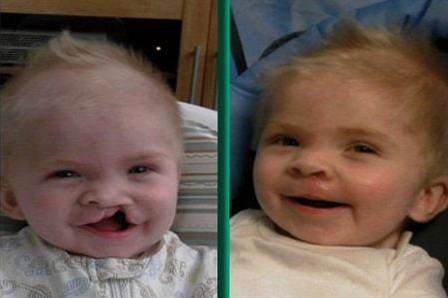
Das DiGeorge-Syndrom, auch als 22q11.2-Deletionssyndrom bekannt, ist eine Krankheit, die durch einen genetischen Defekt verursacht wird und zur Entwicklung verschiedener körperlicher und organischer Missbildungen führt.
In diesem Sinne beruht dieses Syndrom im Wesentlichen auf fehlerhaften Entwicklungsprozessen während der pränatalen oder Schwangerschaftsphase, die hauptsächlich in der 3. und 8. Schwangerschaftswoche auftreten..
Insbesondere in der 5. Schwangerschaftswoche beginnen die embryonalen Strukturen einen Prozess der Bildung und Entwicklung verschiedener Strukturen und Organe (Vera de Pedro et al., 2007)..
So führt eine Gruppe bestimmter Zellen zur Entwicklung des Gesichts, verschiedener Teile des Gehirns, des Thymus, des Herzens, der Aorta und der Nebenschilddrüsen..
Dieses "Feld von Zellen" befindet sich normalerweise um den Bereich oder Bereich hinter dem Hals des Embryos während der Schwangerschaft. Auf diese Weise ist es wichtig, dass sich diese Zellen in Richtung der verschiedenen spezifischen Bereiche für jede Struktur bewegen, damit sich der Rest der Strukturen zu bilden und zu differenzieren beginnt..
In dieser Entwicklungsphase werden die Schleimbeutel, Bögen und Fissuren des Rachens, der Thymusdrüsen und die Nebenschilddrüsen gebildet und später ein Teil der Schädel- und Gesichtsstrukturen oder verschiedene Teile des Bindegewebes.
Auf diese Weise führen die für das DiGeroge-Syndrom typischen genetischen Anomalien zu einer systematischen Veränderung dieses pränatalen Bildungsprozesses, was zu schwerwiegenden Entwicklungsstörungen führt.
Infolgedessen sind die am stärksten betroffenen Gebiete normalerweise:
- Herz: Diese Struktur ist eines der lebenswichtigen Organe für unser Überleben. Es ist Teil des Kreislaufsystems und hat die wesentliche Funktion, Blut in den Rest des Körpers zu pumpen.
- Gesichtskonfiguration: Die Bildung der Gesichtsstruktur hängt von der korrekten Bildung des Schädels, der Augäpfel, des bukkalen Systems, der Ohren usw. ab..
- Betrug: Diese Struktur spielt eine grundlegende Rolle im Immunsystem, da sie für die Reifung von Lymphozyten oder T-Zellen verantwortlich ist.
- Nebenschilddrüsen: Sie bestehen aus einer Reihe von endokrinen Drüsen, die unter anderem eine wichtige Rolle bei der Regulierung von Kalzium spielen.
Somit hängen die am stärksten vom DiGeorge-Syndrom betroffenen Bereiche mit dem Defekt der Embryonalbildung in Bereichen zusammen, die mit dem Hals und angrenzenden Regionen verbunden sind..
Das DiGeroge-Syndrom hat eine geschätzte Prävalenz von 1 Fall pro 4.000 Menschen in der Allgemeinbevölkerung.
Zahlreiche epidemiologische Studien weisen jedoch auf eine höhere Prävalenz hin, die hauptsächlich auf die Heterogenität des klinischen Verlaufs und die Schwierigkeit zurückzuführen ist, eine frühzeitige Diagnose zu stellen.
Darüber hinaus gilt das DiGeorge-Syndrom sowohl in den USA als auch international als eine der häufigsten Ursachen für angeborene Herzfehler und Gesichtsfehlbildungen..
Andererseits wurde in Bezug auf epidemiologische Merkmale soziodemografischer Natur eine Prävalenz von 1 Fall pro 6.000 Personen kaukasischer, asiatischer und afro-abstammender Herkunft festgestellt, während bei Hispanics die Prävalenz einen Fall für beträgt alle 3.800 Personen.
Bei den häufigsten Anzeichen und Symptomen des DiGeorge-Syndroms muss darauf hingewiesen werden, dass es sich um einen klinischen Verlauf mit variabler Ausdruckskraft handelt.
In diesem Fall weisen die medizinischen Komplikationen bei einigen Patienten einen schweren Status auf, der zum frühen Tod führen kann. In anderen Fällen stellen die Merkmale normalerweise einen minimalen Kompromiss für das Überleben und die Funktionalität der betroffenen Person dar..
Daher weisen nicht alle vom Di-George-Syndrom Betroffenen die gleiche Beeinträchtigung auf, sie umfassen jedoch normalerweise eine oder mehrere verwandte Veränderungen.
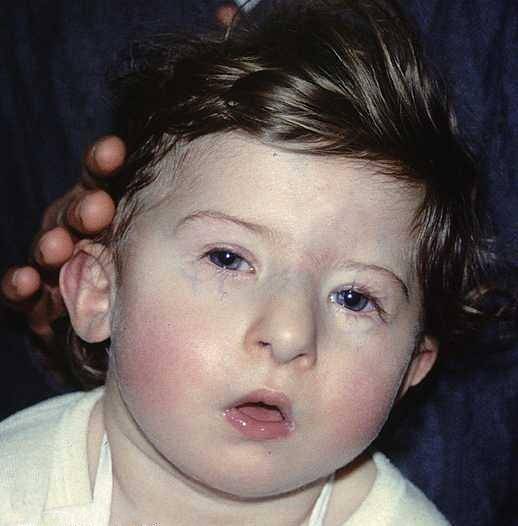
Die Veränderungen in Bezug auf die Gesichtskonfiguration stellen eines der auffälligsten visuellen Merkmale des DiGeorge-Syndroms dar. Im Allgemeinen werden diese definiert durch:
- Mikrozephalie: Der Kopf entwickelt sich mit einer kleineren oder kleineren Dimension als erwartet für den Entwicklungsstand und das chronologische Alter der betroffenen Person. Zusätzlich entwickelt sich normalerweise eine röhrenförmige Nasenstruktur, die von flachen oder schlecht akzentuierten Wangen begleitet wird.
- Unterkieferhyploplasie und Retrognathie: Die Struktur des Kiefers ist nicht voll entwickelt. Daher hat es in vielen Fällen eine reduzierte Größe oder eine veränderte Position, die weiter hinten als gewöhnlich liegt..
- Augenveränderung: Im Allgemeinen neigen die Augen dazu, in Richtung der unteren Ebene eingeschlossen zu sein. Außerdem können Mikrophthalmie (Unterentwicklung eines der Augäpfel), Katarakte (Opazität der Augenlinse) oder Zyanose (blaue Färbung) um die Augen herum auftreten.
- Veränderung der Ohrmuschel: Es ist möglich, eine Asymmetrie in der Konfiguration der Ohren zu identifizieren. Sie haben normalerweise eine geringe Implantation mit Missbildungen in den Lappen und anderen äußeren Bereichen der Ohrmuschel.
- Orale Missbildungen: Die Konfiguration des Mundes zeigt normalerweise ein gewölbtes Erscheinungsbild in Richtung der oberen Ebene, das durch das Vorhandensein eines langen und akzentuierten Sulcus nasolabialis und einer Gaumenspalte gekennzeichnet ist.
Herzanomalien umfassen häufig eine Vielzahl von Defekten. Die am stärksten betroffenen Bereiche hängen jedoch mit der Aorta und den damit verbundenen Herzstrukturen zusammen:
- Septumdefekte: Die Wand oder Struktur, die die Herzkammern trennt, die für das Pumpen von Blut verantwortlich sind, kann unvollständig oder fehlerhaft ausgebildet sein.
- Fehlbildung des Aortenbogens: Verschiedene Anomalien können auch im Aortensegment beschrieben werden, das sich zwischen dem aufsteigenden und dem absteigenden Pfad befindet.
- Fallot-Tetralogie: Diese Pathologie bezieht sich auf das Vorhandensein von Veränderungen im ventrikulären Septumdefekt, eine signifikante Verengung der Lungenarterie, eine abnormale Position der Aorta und eine Verdickung des rechtsventrikulären Bereichs.
Menschen, die vom DiGeorge-Syndrom betroffen sind, sind normalerweise sehr anfällig für verschiedene Arten von Pathologien, die hauptsächlich infektiös sind (Viren, Pilze, Bakterien usw.)..
Diese Tatsache ist auf das Vorhandensein einer Funktionsstörung des Immunsystems aufgrund einer mangelhaften Entwicklung des Typs und der Produktion von Lymphozyten und T-Zellen zurückzuführen.
Das Immunsystem besteht aus einer Vielzahl von Organen, Strukturen, Geweben und Zellen, die uns zusammen vor Umwelt- und inneren Krankheitserregern schützen..
In diesem Sinne führt das DiGeorge-Syndrom zu einer mangelhaften oder unvollständigen Bildung des Thymus, was zu Veränderungen seiner Funktionalität und seiner endgültigen Position führt..
Im Allgemeinen ist die auffälligste Anomalie die Hypofunktionalität von T-Lymphozyten, die für die Produktion von Immunglobulinen und Antikörpern essentiell ist..
In diesem Fall haben Menschen, die vom Digeorge-Syndrom betroffen sind, normalerweise eine ungewöhnlich niedrige Calciumkonzentration im Körper und im Blutkreislauf..
Dieser medizinische Zustand ist im Wesentlichen auf das Vorhandensein von Anomalien in den Nebenschilddrüsen zurückzuführen, die auf eine Unterentwicklung ihrer Bestandteile zurückzuführen sind (PrimaryInmune, 2011)..
Diese Drüsen befinden sich im Nacken und in einer Position nahe der Schilddrüse. In diesem Fall haben sie jedoch ein reduziertes Volumen, so dass dies einen erheblichen Einfluss auf die Kontrolle des Stoffwechsels und des Kalziumhaushalts im Körper hat..
In diesem Fall liegt der Kalziumspiegel im Blut normalerweise unter 2,1-8,5 mm / dl, was zu verschiedenen medizinischen Komplikationen wie Krämpfen, Muskelreizbarkeit, Taubheitsgefühl, Stimmungsschwankungen, kognitiven Defiziten usw. führt..
Zusätzlich zu den oben beschriebenen Anzeichen und Symptomen ist es möglich, andere zu identifizieren, die mit der kognitiven und intellektuellen Sphäre der Betroffenen zusammenhängen..
Insbesondere in diagnostizierten Fällen wurden unter anderem Lernschwierigkeiten, mäßiges intellektuelles Defizit, Aufmerksamkeitsdefizit, Stimmungsschwankungen, Angststörungen beschrieben..
Der genetische Ursprung des DiGeorge-Syndroms ist mit dem Vorhandensein von Veränderungen im Chromosom 22 verbunden, insbesondere an der Stelle 22q11.2. Insbesondere ist dies auf das Fehlen einer DNA-Sequenz zurückzuführen, die aus einer Anzahl von 30 bis 40 verschiedenen Genen besteht..
Obwohl ein großer Teil der beteiligten Gene noch nicht im Detail identifiziert wurde, tritt das Fehlen dieser großen Gruppe in mehr als 90% der Fälle als De-novo-Mutation auf, während etwa 7% auf erbliche Faktoren zurückzuführen sind.
Für die Feststellung der Diagnose des DiGeorge-Syndroms ist es wichtig, die wichtigsten klinischen Anzeichen dieser Pathologie zu identifizieren:
- Gesichtsfehler.
- Herzfehler.
- Immunschwäche.
- Hypokalzämie.
In diesem Sinne ist es neben der Analyse der Krankengeschichte und der körperlichen Untersuchung unerlässlich, verschiedene Labortests wie Echokardiographie, Ultraschall, immunologische Untersuchung und serumanalytische Untersuchungen durchzuführen..
Ein wichtiger Aspekt ist außerdem die genetische Untersuchung, die hauptsächlich durch fluoreszierende In-situ-Hybridisierung (FISH) durchgeführt wird..
Wie wir in der Erstbeschreibung ausgeführt haben, soll die Behandlung hauptsächlich die Anzeichen und Symptome kontrollieren und korrigieren, die durch diese Art von Krankheit verursacht werden..
Im Falle einer Hypokalzämie wird sie normalerweise durch die Verabreichung von Kalzium- und / oder Vitamin-D-Präparaten behandelt..
Andererseits können im Fall von Immunschwäche, obwohl sie dazu neigen, sich mit dem Alter zu verbessern, verschiedene Ansätze verwendet werden, wie die Transplantation eines Teils des Thymusgewebes, die T-Lymphozyten-Therapie oder die Knochenmarktransplantation..
Bei Gesichts- und Mundfehlbildungen werden normalerweise chirurgische Reparaturen durchgeführt, die das physische Erscheinungsbild und die Funktionalität dieser Knochen verbessern.
Schließlich können bei Herzveränderungen beide Medikamente zur Behandlung und Korrektur durch Operation verabreicht werden..
In den meisten Fällen erreichen betroffene Menschen normalerweise das Erwachsenenalter. Ein erheblicher Prozentsatz von ihnen beginnt jedoch, wichtige immunologische und / oder kardiale Anomalien zu entwickeln, die einen vorzeitigen Tod verursachen, insbesondere im ersten Lebensjahr..



Bisher hat noch niemand einen Kommentar zu diesem Artikel abgegeben.