
Das Apert-Syndrom oder Akrozephalosyndaktylie Typ I (ACS1) ist eine Pathologie genetischen Ursprungs, die durch das Vorhandensein verschiedener Veränderungen und Missbildungen in Schädel, Gesicht und Extremitäten gekennzeichnet ist.
Auf klinischer Ebene ist das Apert-Syndrom durch das Vorhandensein oder die Entwicklung eines spitzen oder langgestreckten Schädels, eines versunkenen Gesichtsbereichs mit einer Veränderung der Projektion der Zähne, einer Fusion und eines Verschlusses der Fingerknochen und Gelenke, einer variablen geistigen Behinderung und Sprachstörungen gekennzeichnet , usw..
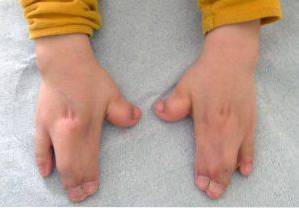
Obwohl diese Pathologie erblich sein kann, tritt das Apert-Syndrom in den meisten Fällen ohne Familienanamnese auf, was im Wesentlichen auf eine De-novo-Mutation während der Schwangerschaftsphase zurückzuführen ist..
Die genetischen Mechanismen, die das Apert-Syndrom verursachen, sind nicht genau bekannt. Gegenwärtig wurden verschiedene genetische Veränderungen identifiziert, die diese Pathologie hervorrufen können, die im Wesentlichen mit Mutationen im FGFR2-Gen zusammenhängen.
Andererseits beginnt die Diagnose des Apert-Syndroms normalerweise mit einem klinischen Verdacht in der pränatalen Phase nach der Identifizierung von Anomalien bei routinemäßigen Ultraschalluntersuchungen und wird durch die Durchführung einer genetischen Studie bestätigt.
In Bezug auf die Behandlung gibt es keine Art von kurativer Intervention für das Apert-Syndrom. Im Laufe der Geschichte dieser Pathologie wurden jedoch verschiedene spezifische Interventionen entwickelt, die üblicherweise Neurochirurgie, kraniofaziale Chirurgie, maxillofaziale Chirurgie, medikamentöse Behandlung, physikalische Therapie, psychologische und neuropsychologische Intervention umfassen.
Artikelverzeichnis
Das Apert-Syndrom ist eine genetische Pathologie, die durch das Vorhandensein verschiedener Skelettfehlbildungen auf Schädel-, Gesichts- und / oder Extremitätenebene gekennzeichnet ist.
Die wesentliche Veränderung des Apert-Syndroms besteht in einem vorzeitigen oder frühen Schließen der Schädelrisse, was zu einem abnormalen Wachstum der übrigen Strukturen von Gesicht und Schädel führt. Zusätzlich zu diesen können auch Missbildungen in den oberen und unteren Extremitäten auftreten, wie z. B. die Verschmelzung von Fingern und Zehen..
Andererseits können auch die kognitiven Fähigkeiten von Menschen mit Apert-Syndrom mit einem variablen Schweregrad von leicht bis mittelschwer beeinträchtigt werden.
Obwohl Baumgartner (1842) und Wheaton (1894) die ersten Erwähnungen zu dieser Krankheit machten, beschrieb der französische Facharzt Eugene Apert dieses Syndrom erst 1906 genau und veröffentlichte den ersten klinischen Bericht.
In seiner Veröffentlichung beschreibt Eugene Apert eine Reihe neuer Fälle von Patienten, die von einem genau definierten Missbildungsmuster betroffen sind und durch die charakteristischen Anzeichen und Symptome dieser Pathologie gekennzeichnet sind..
So wurden erst 1995 die ätiologischen genetischen Faktoren des Apert-Syndroms identifiziert. Insbesondere beschrieben Wilkie et al. Das Vorhandensein von zwei Mutationen im FGFR2-Gen bei etwa 40 betroffenen Patienten..
Darüber hinaus ist das Apert-Syndrom eine Erkrankung, die in die Krankheiten oder Pathologien eingeteilt wird, die durch Craniosynostose (vorzeitiger Verschluss von Schädelnähten) gekennzeichnet sind..
Andere Pathologien, die zu dieser Gruppe gehören, sind das Pfeiffer-Syndrom, das Crouzon-Syndrom, das Saethre-Chotzcen-Syndrom und das Carpenter-Syndrom..
Das Apert-Syndrom wird als seltene oder seltene Pathologie angesehen, dh es hat eine Prävalenz von weniger als einem Fall pro 15.000 Einwohner der Allgemeinbevölkerung..
Insbesondere tritt das Apert-Syndrom bei einer Person pro 160.000 bis 200.000 Geburten auf, und außerdem besteht eine 50% ige Wahrscheinlichkeit, diese Pathologie auf erblicher Ebene zu übertragen.
Darüber hinaus wurde in Bezug auf die Verteilung nach Geschlecht weder eine höhere Prävalenz bei Männern oder Frauen festgestellt, noch wurde sie mit bestimmten ethnischen Gruppen oder geografischen Orten in Verbindung gebracht..
Derzeit und angesichts der Tatsache, dass das Apert-Syndrom ungefähr 1984 in klinischen Berichten und in der medizinischen Literatur identifiziert wurde, die mehr als 300 Fälle dieser Pathologie veröffentlicht haben.
Die klinischen Manifestationen des Apert-Syndroms umfassen normalerweise eine Fehlbildung oder unvollständige Entwicklung der Schädelstruktur, einen atypischen Phänotyp oder ein Gesichtsmuster und Skelettveränderungen in den Extremitäten..
Beim Apert-Syndrom hängt die zentrale Beteiligung mit der Bildung und dem Verschluss der Knochenstruktur des Schädels zusammen. Während der Embryonalentwicklung tritt ein Prozess namens Creneosynostose auf, der durch vorzeitiges Schließen der Schädelnähte gekennzeichnet ist.
Schädelrisse oder -nähte sind eine Art von faserigen Gewebebändern, die das grundlegende Ziel haben, die Knochen, aus denen der Schädel besteht, zu verbinden (frontal, okzipital, parietal und temporal)..
Während der Schwangerschaftsphase und der frühen postnatalen Phase wird die Knochenstruktur, aus der der Schädel besteht, dank dieser faserigen und elastischen Gewebe zusammengehalten.
Normalerweise verschmelzen die Schädelknochen erst nach 12 bis 18 Monaten. Das Vorhandensein von Schwachstellen oder Zwischenräumen zwischen den Schädelknochen ist Teil der normalen kindlichen Entwicklung.
Während des gesamten Kindheitsstadiums ermöglichen diese Nähte oder flexiblen Regionen dem Gehirn ein beschleunigtes Wachstum und schützen es zusätzlich vor Stößen.
Daher macht beim Apert-Syndrom ein vorzeitiger Verschluss dieser Schädelnähte und Schädelknochen eine normale Entwicklung des Schädel- und Gehirnwachstums unmöglich..
Folglich können die häufigsten Anzeichen und Symptome des Apert-Syndroms sein:
Diese Arten von Veränderungen betreffen hauptsächlich die oberen und unteren Extremitäten, normalerweise die Verschmelzung und Entwicklung der Finger..
Darüber hinaus können auch andere klinische Befunde auf muskuloskelettaler Ebene, Verkürzung verschiedener Knochen (Radius, Humerus, Femur), Hypoplasie des Schulterblatts oder des Beckens, Fusion von Halswirbeln beobachtet werden.
Infolgedessen weisen viele Betroffene eine verminderte Beweglichkeit der Gelenke auf und können daher verschiedene Schwierigkeiten beim Erwerb von Brutto- und Feinmotorik entwickeln.
Diese Arten von Anomalien sind bei betroffenen Personen sehr heterogen und variabel. Einige der häufigsten wurden jedoch identifiziert:
Die ätiologische Veränderung dieser Pathologie kann zur Entwicklung von Läsionen oder sekundären Pathologien auf morphologischer und struktureller Ebene in verschiedenen Bereichen des Körpers führen, von denen einige Folgendes umfassen:
Trotz der Tatsache, dass in vielen Fällen das Vorhandensein einer allgemeinen Veränderung der kognitiven Funktionen und des intellektuellen Niveaus beobachtet werden kann, ist eine geistige Behinderung nicht in allen Fällen des Apert-Syndroms eindeutig vorhanden..
Darüber hinaus kann dies in Fällen, in denen eine Beeinträchtigung des intellektuellen Niveaus vorliegt, auf einer Skala von leicht bis mäßig variabel sein.
Andererseits ist im sprachlichen Bereich die Entwicklung verschiedener Defizite häufig, hauptsächlich im Zusammenhang mit der Artikulation von Geräuschen infolge von Fehlbildungen des Unterkiefers und des Mundes..
Das Apert-Syndrom ist auf das Vorhandensein einer spezifischen Mutation im FGFR2-Gen zurückzuführen. Experimentelle Studien haben gezeigt, dass dieses Gen für die Produktion eines Proteins verantwortlich ist, das als Fibroblasten-Wachstumsfaktor-Rezeptor 2 bezeichnet wird.
Unter den Funktionen dieses Faktors wird das Senden verschiedener chemischer Signale an unreife Zellen beschrieben, um deren Transformation und Differenzierung in Knochenzellen während der fetalen oder pränatalen Entwicklungsphase zu bewirken..
Daher verändert das Vorhandensein von Mutationen im FGFR2-Gen die Funktion dieses Proteins und kann daher die frühe Fusion der Knochen von Schädel, Hand und Füßen verursachen..
Ein großer Teil der klinischen Merkmale des Apert-Syndroms kann während der Schwangerschaft identifiziert werden, insbesondere bei Ultraschalluntersuchungen der Schwangerschaft und der Entwicklung des Fötus..
Wenn also ein klinischer Verdacht besteht, wird eine genetische Studie erneut gestartet, um das Vorhandensein einer genetischen Mutation zu identifizieren, die mit dem Apert-Syndrom kompatibel ist..
Wenn andererseits die Anzeichen subtil sind oder vor der Geburt nicht identifiziert wurden, ist es danach möglich, eine detaillierte physikalische Analyse und verschiedene Gentests durchzuführen, um die Diagnose zu bestätigen..
Obwohl es keine spezifische Heilung für das Apert-Syndrom gibt, wurden verschiedene Ansätze zur Behandlung der Symptome und medizinischen Komplikationen dieser Pathologie beschrieben..
Die wirksamsten therapeutischen Interventionen sind solche, die frühzeitig in den ersten Augenblicken des Lebens durchgeführt werden und an denen Fachleute aus verschiedenen Bereichen beteiligt sind.
In der Regel erfordert die Behandlung betroffener Kinder eine individuelle Planung, wobei mehrere Operationen geplant sind. Daher basiert das Management dieser Pathologie auf der Korrektur von Skelett- und Schädel-Gesichtsfehlbildungen sowie auf psychologischer und neuropsychologischer Unterstützung..
Ziel der Neurochirurgie ist es, das Schädelgewölbe zu rekonstruieren, während Spezialisten für Kiefer- und Gesichtschirurgie versuchen, Gesichtsfehlbildungen zu korrigieren. Andererseits ist auch die Teilnahme von Unfallchirurgen zur Rekonstruktion der in Händen und Füßen vorhandenen Missbildungen häufig.
Darüber hinaus ist die Gestaltung individueller Programme zur frühzeitigen Stimulation, zur Rehabilitation der Kommunikation, zum Training sozialer Kompetenzen oder zur psychopädagogischen Nachsorge von Vorteil, um eine optimale, funktionale und unabhängige Entwicklung der betroffenen Personen zu erreichen..



Bisher hat noch niemand einen Kommentar zu diesem Artikel abgegeben.